 Research Article
Research Article





Abstract
X-linked hypohidrotic ectodermal dysplasia (XLHED) can be characterized by hypohidrosis, sparse hair, hypodontia, and characteristic facial features and usually caused by mutations of ectodysplasin A (EDA) gene located on the X chromosome. In this study, we examined a HED pedigree and carried out molecular biological experiments of the disease. A novel nonsense mutation was revealed by direct sequencing analysis in the EDA exons (c.526 G>T). The function of the mutant EDA gene was subsequently confirmed by in vitro study in human embryonic kidney 293T cells transfected with mutant or wild isoforms of EDA. The mutant-type EDA1 sequence showed total loss of the capability of protein producing compared with wild-type EDA1. This novel nonsense EDA mutation was considered to be the cause of HED in the pedigree reported here. Our findings, combined with those reported elsewhere, provide an improved understanding of the pathogenic mechanism of HED as well as important information for a genetic diagnosis.
Keywords: Etodysplasin A; Hypohidrotic Ectodermal Dysplasia; Mutation; Nonsense
Abbreviations:XLHED: X-Linked Hypohidrotic Ectodermal Dysplasia; EDA: Ectodysplasin A; HED: Hypohidrotic Ectodermal Dysplasia; SNP: Single Nucleotide Polymorphisms; TNF: Tumor Necrosis Factor; PCR: Polymerase Chain Reaction; PTC: Premature Termination Codon
Introduction
Ectodermal dysplasia (ED) contains a group more than 170 types of congenital disorders resulting from abnormal development of ectoderm-derived structures [1,2]. The most common type of ED, hypohidrotic ectodermal dysplasia (HED), occurs in 1 in 10,000–100,000 births [3,4]. HED is characterized by hypohidrosis, hypotrichosis, hypodontia, and characteristic facial features. The common clinical signs include dry skin, sparse hair, deformity teeth, diminishing or absence of eccrine function and characteristic facial features including frontal bossing, chin prominence, saddle nose, maxillary hypoplasia, low-set ears, wrinkles, and periorbital hyperpigmentation. Three inheritance patterns of HED are known: X-linked recessive, autosomal recessive, and autosomal dominant, corresponding to the main causative genes (EDA, EDAR and EDARADD). The X-linked HED (XLHED) is caused by mutations of ectodysplasin A (EDA) gene (Genbank accession number: NM 001399) located within the X q12-q13.1 region, causing an X-linked recessive pattern of inheritance [5,6].
The hemizygous males are often most severely affected, showing the “classic” clinical features, while heterozygous females generally show normal or moderately affected features [3-7]. The EDA-A1 protein which contains 391 amino acids, is the most common and also the largest product of the EDA gene [8]. It is a trimeric type II transmembrane protein composed of a transmembrane domain, a furin protease recognition site, a 19 repeat Gly-X-Y collagenous domain as well as a tumor necrosis factor (TNF) homology C-terminal domain [9]. To date, more than 300 mutations of the EDA gene have been reported [10], including missense, nonsense and splice mutations as well as small deletions [11,12]. 11 of the 16 nonsense mutations reported in the database of single nucleotide polymorphisms (SNP) of the National Center for Biotechnology. Information (NCBI) are described as “pathogenic”, although none has been validated in functional studies. In this study, we describe the clinical features and molecular characterization of a novel nonsense mutation identified in a Chinese family with XLHED. We also performed in vitro functional experiments to elucidate the molecular mechanisms underlying the effects of this mutation.
Materials and Methods
Subjects
The proband were referred to the School and Hospital of Stomatology, Peking University (China) for oligodontia. After examination by pediatric dentistry specialists, the proband were diagnosed as XLHED. His guardian provided informed consent to the genetic studies. This study has been approved by Peking University school of stomatology institutional review board(approval number: PKUSSIRB-201628059). The proband was a 1-year-7-monthold boy who presented with absence of several teeth. Physical examination showed apparently dry skin with hypohidrosis, sparse hair and eyebrows, but normal nails. Other facial features including frontal bossing, chin prominence, maxillary hypoplasia and protuberant lips were also obvious in this patient. Intraoral examination revealed only two cone-shaped upper anterior teeth, a lack of alveolar bone development and ordinary salivary flow. Similar phenotype in his family members was denied. The pedigree diagram is shown in (Figure 1).
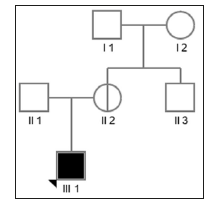
Figure 1: Pedigree of the family with an X-linked recessive inheritance pattern. Affected individuals are represented by filled squares (male), while non-penetrant carriers are represented by circles with a vertical line (female).
Mutation Analysis
Blood samples were obtained from the proband and his family members. Genomic DNA was extracted from peripheral blood leukocytes using TIANamp Blood DNA Kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. DNA sequences were amplified by polymerase chain reaction (PCR) using specific primers covering the exons and intron–exon boundaries of the EDA gene as described previously [5]. Sanger sequences of individual genes were analyzed using the Chromas chromatogram viewer and the National Center for Biotechnology Information Basic Local Alignment Search Tool (NCBI BLAST, The detected variant was assessed with the mutation prediction tool Mutation Taster.
Construction Of Eda Expression Vectors
Mammalian expression vectors for secreted Flag-tagged forms of wild-type EDA1 and EDA1 with HED-causing mutation G176X. EDA1 with HED-causing mutation H252L and non-syndromic tooth agenesis-causing mutations S374R were also constructed to make a contrast. Construction of above expression vectors was performed by GENEWIZ, Suzhou, China. All constructs were verified by direct sequencing.
Expression of Soluble Eda in Cells and Supernatants
Vectors encoding soluble wild-type or mutant Flag-tagged EDA1 protein were transfected into human embryonic kidney 293T cells. The cells were maintained in serum-free OptiMEM (Invitrogen Corporation, Carlsbad, CA, USA) culture medium for 48 hours. Subsequently, the cells and supernatants were collected separately. The detection of Flag-tagged EDA1 proteins in cell lysates and supernatants was then performed by anti-Flag western blot.
Results
Direct sequencing revealed the presence of a previously unidentified mutation in the exon 3(c.526G>T, p. G176X) in the proband. The mother of the proband carried the same mutation. This mutation was not found in the healthy members of the family (Figure 2). The selected EDA-A1 mutants H252L and S374R affecting the TNF homology domain could be expressed as soluble, Flag-tagged secreted proteins but mutant G176X led to entire failure of detecting Flag-tagged secreted proteins either in the cell lysate or in the supernatant (Figure 3).
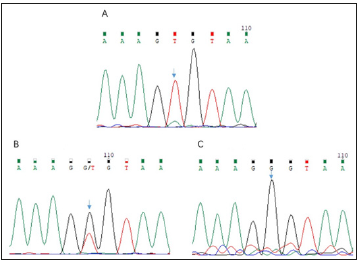
Figure 2: A: Sequencing chromatogram of the splicing mutation c.526G>T in the EDA gene of the proband. B: Sequencing chromatogram of the heterozygous mutation c.526G>T in the EDA gene of the female carrier. C: Sequencing chromatogram of the EDA gene of the father.
Discussion
The transmembrane protein ectodysplasin A is expressed by keratinocytes, hair follicles, and sweat glands. The TNF-related ectodysplasin A pathway plays an important role in embryonic development and ectodermal structure formation [13]. As a TNFrelated ligand, it has been confirmed that EDA mutations are responsible for X-linked HED. The human EDA gene contains 12 exons, with five different transcripts of the EDA protein produced by alternative splicing according to the GenBank database. Among them, the EDA-A1 transcript, which contains eight exons, is the longest and most abundant product generated by transcription of the EDA gene. Here, we identified a pedigree with HED carrying a novel mutation in the exon 3 c.526G>T in EDA gene. It leads to a premature transcription termination codon of EDA-A1 DNA sequence at the 176th amino acid and a truncated protein product in theory. However, as the anti-Flag western blot experiment revealed, the G176X mutant plasmid failed to express detectable secreted protein product either in the cell lysate or in the supernatant (Figure 3).
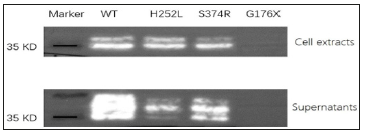
Figure 3: Expression of wild and mutant types of EDA-A1. Cell extracts and supernatants of transfected cells are shown in the two panels. H252L mutation is a HED related mutation reported previously, while S374R is a hypodontia related mutation reported previously. Nonsense mutant G176X led to entire failure of detecting Flag-tagged secreted proteins either in the cell lysate or in the supernatant.
The mechanism of this phenomenon could be attributed to a pathway called “Nonsense-Mediated mRNA Decay (NMD)” [14,15]. Nonsense mutations in genes would lead to the production of the premature termination codon (PTC). To avoid the translation of potential harmful C-terminal truncated protein product from the mRNA containing the PTC, the NMD pathway acting as a quality control mechanism of mRNA is responsible for the recognition and degradation of such abnormal mRNA. Similar phenomenon was observed in the beta-globin gene region of thalassemia patients as early as in the 1980s and the mechanism of NMD pathway has been studied and proved widely till now [16]. In conclusion, we identified a novel mutation in the EDA exon (c.526G>T, p.G176X), and demonstrated its role in resulting in failure of expressing detectable secreted protein product. Thus, we hypothesize that this mutation is the cause of HED in the patient described here.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 81500828].
Acknowledgment
We are grateful to all the patients and their family members for their participation in this study
References
- Pinheiro M, Freire-Maia N (1994) Ectodermal dysplasias: a clinical classification and a causal review. Am J Med Genet 53: 153-162.
- Priolo M, Lagana C (2001) Ectodermal dysplasias: a new clinical-genetic classification. J Med Genet 38: 579-585.
- Itin PH, Fistarol SK (2004) Ectodermal dysplasias. Am J Med Genet 131(1): 45-51.
- Chassaing N, Bourthoumieu S, Cossee M, Calvas P, Vincent MC, et al. (2006) Mutations in EDAR account for one quarter of non-ED1-related hypohidrotic ectodermal dysplasia. Hum Mutat 27: 255-259.
- Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, et al. (1996) X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 13: 409-416.
- Deshmukh S, Prashanth S (2012) Ectodermal Dysplasia: A Genetic Review. Int J Clin Pediatr Dent 5(3): 197-202.
- Clarke A, Phillips DI, Brown R, Harper PS (1987) Clinical aspects of X-linked hypohidrotic ectodermal dysplasia. Arch Dis Child 62: 989-996.
- Podzus, J Kowalczyk-Quintas C, Schuepbach-Mallepell S, Willen L, Staehlin G, et al. (2017) Ectodysplasin A in Biological Fluids and Diagnosis of Ectodermal Dysplasia. J Dent Res 96(2): 217-224.
- Monreal AW, Zonana J, Ferguson B (1998) Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am J Hum Genet 63: 380- 389.
- Trzeciak WH, Koczorowski R (2016) Molecular basis of hypohidrotic ectodermal dysplasia: an update. J Appl Genet 57: 51-61.
- Cluzeau C, Hadj-Rabia S, Jambou M, Mansour S, Guigue P, et al. (2011) Only four genes (EDA1, EDAR, EDARADD and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum Mutat 32(1): 70-72.
- Maquat LE (1981) Unstable beta-globin mRNA in mRNA-deficient beta o thalassemia. Cell 27(3): 543-553.
- Stalder L, O Muhlemann (2008) The meaning of nonsense. Trends Cell Biol 18(7): 315-321.
- Mikkola ML, Thesleff I (2003) Ectodysplasin signaling in development. Cytokine Growth Factor Rev 14(3-4): 211-224.
- Brogna S, J Wen (2009) Nonsense-mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol 16(2): 107-113.
- Kan YW (1980) Polymorphism of DNA sequence in the beta-globin gene region. Application to prenatal diagnosis of beta 0 thalassemia in Sardinia. N Engl J Med 302(4): 185-188.
