 Review Article
Review ArticleAbstract
The 1,2-bis(sulfonyl)hydrazines (BSHs) have very short tunable half-life values, which give them unmatched abilities in terms of their activity confinement to the target site. Furthermore, their therapeutic cytotoxicity, which is delivered as a consequence of their decay, is due to the alkylation of the O-6 position of guanine. This has led to the suggested use of BSHs in tumor targeted applications, in particular to target the subset of tumors lacking O6-methylguanine-DNA methyltransferase (MGMT), the sole enzyme involved in the repair of guanine O-6 alkyl lesions. While current BSHs largely owe their therapeutic cytotoxicity to the alkylation of the O-6 position of guanine, there is evidence that alkylation does occur at multiple sites in DNA, and in other biomolecules, as a consequence of alternative decomposition pathways that lead to the production of electrophiles with a preference for other nucleophilic sites. In this paper we discuss strategies to minimize these alternative reaction pathways, thereby further increasing the activity and selectivity of all categories of BSH prodrugs.
Introduction
The DNA repair protein O6-methylguanine-DNA
methyltransferase (MGMT) is either absent or expressed in
insignificant levels in a small sub-set of many tumor types [1,2]. The
size of this subpopulation has been reported to be between 5-30%
depending upon the tumor type [1-5]. DNA damage at the O-6
position of DNA guanine is unusual as it relies solely upon MGMT
for its repair [1]. Thus, the loss of MGMT renders cells completely
vulnerable to guanine O-6 assaults. Each MGMT molecule can only
repair a single guanine O-6 lesion as MGMT is a ‘suicide enzyme’ [1].
Once a cell’s MGMT reserves are exhausted, DNA guanine O-6 lesion
repair ceases until fresh MGMT is synthesized [6] (a relatively slow
process). Therefore, cells lacking MGMT have an exploitable hole
in their DNA repair machinery. Neoplasms with this targetable
vulnerability may be identified by direct assays to determine MGMT
activity, or the lack of MGMT activity can be inferred based on
methylation-specific PCR of the MGMT gene promoter. These assays
may allow the pre-selection of patients for clinical trials with a high
probability of responding to agents attacking the O-6 position of
DNA guanine and assist in the attainment of personalized medicine.
Anticancer drugs are generally approved on a disease-by-disease
basis, but the presence of common therapeutic susceptibilities in
diverse tumor types is likely to change this approach [7].
Clinical chemotherapeutic alkylators such as the
chloroethylnitrosoureas (CNUs) generate a more diverse array of
toxic electrophiles than chloroethylating BSHs. This results in CNUs
producing a wider range of lesion types at many nucleophilic sites
within DNA, and a smaller proportion of guanine O-6 lesions [8-12].
Thus, with the CNUs there is considerably more MGMT-independent
toxicity to normal cells [3]. Herein, we describe the design of novel
DNA cross-linking 1,2-bis(sulfonyl)hydrazines (BSHs) optimized to
target the O-6 position of DNA guanine to a further enhanced level,
such that they essentially owe the entirety of their tumor-selective
cytotoxicity to the generation of guanine O-6 lesions. These
improved agents would be expected to cause DNA damage that is
highly lethal to MGMT-deficient tumors but is readily repaired by
normal cells, resulting in minimal toxic side effects. Currently, there are no approved agents with sufficient DNA guanine O-6 target
specificity and potency to effectively accomplish this aim. The
proposed improved BSHs can be readily incorporated into a variety
of tumor-specific delivery platforms for additional tumor selectivity
[2,5]. By exceeding the distinct protection limits provided by MGMT
[2,13], tumor-targeted BSHs are expected to function against both
moderate MGMT- expressing and non-expressing tumors. Normal
tissue MGMT levels would afford protection from the lesser
exposures received due to active agent leakage and non-targeted
delivery. In targeted applications, the highly tunable half-life of
the liberated BSH is also a major advantage since it can be used to
limit the spatial volume surrounding the activating cell or region
to which the cytotoxic stress is delivered, minimizing off-target
cytotoxicity due to leakage from the intended target site [14].
Using existing therapeutics in targeted applications is inherently flawed as their utility as ‘free-agents’ is due to pharmacokinetic properties that favor rapid distribution and tissue penetration, and these very same properties will enable the escape and systemic distribution of a formerly targeted agent [14-16]. The unique properties of BSHs circumvent these limitations and advocate their further development and incorporation into tumor specific delivery platforms [14]. Currently, there are two basic types of approved agents that alkylate the O-6 position of DNA guanine: those which methylate and those that chloroethylate. Unfortunately, the O6-methylguanine lesion is of very low cytotoxicity even in sensitive MGMT-deficient cells, since ~ 5600 lesions/cell are required for a 50% growth inhibition [13], and loss of MMR essentially negates the toxicity of this lesion [17-21]. Despite this limitation, the guanine O-6 methylating agent, temozolomide is likely the most efficacious current agent against MGMT-deficient tumors. Guanine O-6 chloroethylators behave differently since the initial lesions formed transition into 1-(N3-cytosinyl),-2-(N1-guaninyl)ethane DNA-DNA interstrand cross-links (G-C ethane cross-links) via an N1,O6-ethanoguanine intermediate [22-24]. While the O6-chloroethylguanine and N1,O6-ethanoguanine lesions can be repaired by MGMT, the highly lethal G-C ethane cross-link cannot [22,25]. Thus, for a cell to survive, the MGMT activity must be large enough to produce a repair rate that clears the cross-link precursors before a small but lethal number transition to cross-links (<10 in some clonal survival assays) [23].
A large excess of MGMT relative to the number of O-6 guanine lesion number is therefore required, but little MGMT depletion occurs at cytotoxic doses. This contrasts with O6-methylguanine where complete MGMT titration has to occur before toxicity is manifested [13,23]. In addition, loss of MMR does not result in resistance to guanine O6-chloroethylation [26]. Cells can repair a very limited number of G-C ethane cross-links using homology directed repair (HDR) and cells lacking both HDR and MGMT are hypersensitive to agents of this type [27]. The higher potency of guanine O-6 chloroethylators (~20-fold) also makes them more attractive than guanine O6-methylators for incorporation into tumor delivery platforms, which are frequently limited in their drug loading capacity [28]. Unfortunately, the relatively non-selective chloroethylnitrosoureas (CNUs), are currently the only clinically approved agents that generate O6-chloroethylguanine. Furthermore, CNUs generate a number of therapeutically irrelevant electrophiles via decomposition routes that are difficult to control or manipulate by structural modification or analog synthesis. This greatly reduces the MGMT dependency of their overall cytotoxicity [8-12]. BCNU, for example, decomposes in a multifarious manner generating chloroethylating, hydroxyethylating, vinylating, aminoethylating and carbamoylating species with a wide range of nucleophile preferences. In addition, BCNU itself may be able to directly alkylate DNA at soft sites such as guanine N-7 [29,30]. This results in BCNU’s cytotoxicity exhibiting a relatively small MGMT dependency of 2- to 3-fold in tissue culture and MGMT knockout mice [13,30-33]. Thus, neither currently approved methylators nor currently approved chloroethylators are ideally suited for specifically targeting MGMT deficiency. Several BSHs come significantly closer to fulfilling the ideal guanine O-6 alkylator requirements, and recent studies on the decomposition pathways open to BSHs have pointed the way to further improving these molecules by precisely tuning them for this specific task [34].
BSHs as O-6 DNA Guanine Targeting Agents
Laromustine (also known as cloretazine, onrigin, VNP40101M, and 101M), a chloroethylating BSH prodrug, was originally designed in our laboratory to eliminate many of the non-efficacious alkylating species generated by the CNUs while retaining the production of the therapeutic electrophiles [29,34,35]. Therefore, hydroxyethylating, vinylating, aminoethylating and direct alkylating activities present in the BCNU were removed, while the generation of oxophilic chloroethylating species favoring the chloroethylation of guanine O-6 (unequivocally required for anticancer activity) and the carbamoylating activities were retained [34,35]. Carbamoylating activity was retained since it had been speculated by several groups that the intracellular release of isocyanates played a role in modulating the biological activity of the CNUs against some specific tumor types [36-42]. Thus, laromustine was significantly less toxic than BCNU towards mice, yet displayed excellent antitumor activity against MGMT-deficient tumor models, producing 100% cures in many cases [36]. Furthermore, laromustine has a therapeutic index (LD50/ED50) against the L1210 leukemia of > 8, more than double that of the best of over 300 nitrosoureas evaluated [36,42]. Although laromustine was the subject of several clinical trials, it was never tested against tumor sub-sets prescreened for low MGMT activity, where a very high response rate would be expected. However, in one Phase I study in patients with refractory leukemia, the MGMT levels in total peripheral blood mononuclear cells (a mixture of peripheral leukemic blast and normal peripheral blood mononucleated cells) were ascertained, and lower MGMT levels were found in responding patients. Median MGMT levels in responding and non-responding patients were 5.73 and 9.21 fmol/ μg of cellular DNA, respectively [43]. A much stronger correlation with therapeutic activity would have been likely if purified leukemic blasts had been assayed rather than total peripheral blood mononuclear cells. Laromustine as a single agent produced an objective response rate (CR, complete remission + CRp, all of the criteria for a CR with platelets less than 100,000/μl) of 32% in de novo AML of poor-risk elderly patients [44]. The Oncologic Drugs Advisory Committee (ODAC) recommended that a successful randomized controlled trial would be required prior to laromustine approval. The developing company (Vion Pharmaceuticals) failed financially at the end of 2009 and trials consistent with the ODAC recommendations were never conducted. This financial failure also resulted in the abandonment of two promising hypoxia targeted BSH prodrugs [45,46] based on 90CE (KS119, KS119W), which had been short-listed for clinical development.
While laromustine exhibits striking activity against neoplasms with absent or very low levels of MGMT its differential toxicity against this subset is still compromised by several readily correctable factors. Laromustine is a simple prodrug, spontaneously activated by base-catalyzed elimination to yield 90CE (the chloroethylating component) and methyl isocyanate (the carbamoylating component). Studies of analogs with segregated chloroethylating and carbamoylating activities [47] indicate that in in vitro experiments utilizing L1210 leukemia wild-type and MGMT transfected L1210 cells, the isocyanate moiety does not contribute to MGMT-dependent differentials in cytotoxicity but does result in non-specific toxicity. This accounts for the lower MGMT-dependent cytotoxicity differential seen with laromustine compared to 90CE [32]. An obvious approach to improving the therapeutic index against MGMT-deficient tumors is to replace the methylaminocarbonyl moiety (the source of carbamoylating activity) of laromustine with a cleavable group that does not give rise to a cytotoxic product with no therapeutic benefit. Some earlier 1,2,2-tris(sulfonyl)-1-(2-chloroethyl)hydrazines (possessing significantly poorer pharmacokinetics) synthesized in our laboratory already fulfilled this condition and produced 100% cures of the L1210 leukemia with less host toxicity as judged by bodyweight loss [48]. However, the major thrust of this paper is to optimize the 90CE moiety itself. An improved 90CE type ‘warhead’ can then be exploited by its incorporation into a wide number of agents from spontaneously activated prodrugs such as laromustine [36] to far more complex tumor-selective delivery platforms [2,5,14].
Alkylating agents generate electrophilic species which react
with biological nucleophiles, including sites within DNA. These
electrophiles can exhibit significant selectivity for their sites of
reaction [49]. However, this selectivity is frequently overlooked in the biological literature where the tendency is to assume that
all alkylating agents are reactive species that uniformly lack
discrimination in their choice of biological nucleophilic targets.
The concept of ‘hard’ and ‘soft’ was introduced as a qualitative
predictor of electrophile/nucleophile reaction preference [49].
Hard electrophiles have a high positive charge density, and tend to
react via SN1 reaction mechanisms with hard nucleophiles which
have a high negative charge density. In contrast, soft electrophiles
have a low charge density or are easily polarized and tend to react
via SN2 reaction mechanisms with soft nucleophiles that have a
low negative charge density or are easily polarized. The activation
energy for reaction is the lowest between pairs of electrophiles and
nucleophiles with closely matching degrees of hardness/softness
[49]. Thus, the relative degree of hardness/softness of electrophiles
determines the range of preferred nucleophiles for reaction. The
extremely wide range of sites alkylated in DNA by BCNU, ranging
from the softest DNA site (N-7 position of guanine) to the hardest
(the negatively charged oxygens of the DNA backbone phosphates)
is more a function of the wide range of disparate electrophilic
species generated by BCNU rather than the nucleophilic promiscuity
of individual electrophilic species [29,49].
Recent studies on the fate of 90CE in different buffer
environments have revealed a surprising catalytic effect of
Brønsted-Lowry bases on the decomposition pathways taken by
90CE [34] and related compounds. Phosphate and its mono- and
diesters were found to be particularly active in this regard [34]. This
Brønsted-Lowry base-catalyzed pathway leads to the production
of major quantities of a relatively soft unwanted thiophilic, and
aromatic nitrogen preferring electrophile [34]. Furthermore, our
studies concerning these decomposition mechanisms/routes have
indicated how to more precisely tune 90CE analogs to:
A. Reduce the production of this unwanted relatively soft
electrophile which diverts material from the desired
decomposition pathway and likely contributes to MGMTindependent
cytotoxicity.
B. More precisely tune the selectivity of the primary
haloethylating/pseudohaloethylating species to target the O-6
position of guanine.
C. Diminish fragmentation of the primary haloethylating/
pseudohaloethylating species to form harder charged
electrophiles that favor the alkylation of the phosphate groups
of the DNA sugar/phosphate backbone.
D. Increase the relative MGMT interceptability of the initial O-6
guanine lesions.
Improvements in any of these parameters will further amplify the strong inverse relationship between MGMT activity and cytotoxicity seen in 90CE analogs, leading to lower host toxicities at cytocidal concentrations towards tumor cells lacking MGMT activity. Prodrug delivery systems with uptake/activation mechanisms based upon nanoparticle-enhanced permeability and retention (EPR) effects [5], hypoxia-specific net reduction [2,14], glutathione S-transferase-catalyzed thiolysis [50], acid-catalyzed activation by solid tumor extracellular acidity [47], and extracellular matrix protease activation [51], can then be used to more selectively deliver improved 90CE analogs and add a further degree of tumor selectivity. Thus, improvements in 90CE type moieties can bring about a wide range of benefits to the design of BSH prodrugs. When coupled with delivery strategies these moieties can display activity against MGMT-expressing tumors by overwhelming their MGMT levels [52,53]. In addition, tumor-targeted prodrugs of short-lived methylating BSH can be used to selectively sensitize tumor cells by ablating tumor MGMT [2,52]. These agents have a significant advantage over equivalently targeted O6-benzylguanine prodrugs in terms of the diminished escape of the activated agent from the target site due to their short half-lives [2,52].
The contribution of Different Decomposition Routes to Efficacy and Toxicity of 90CE
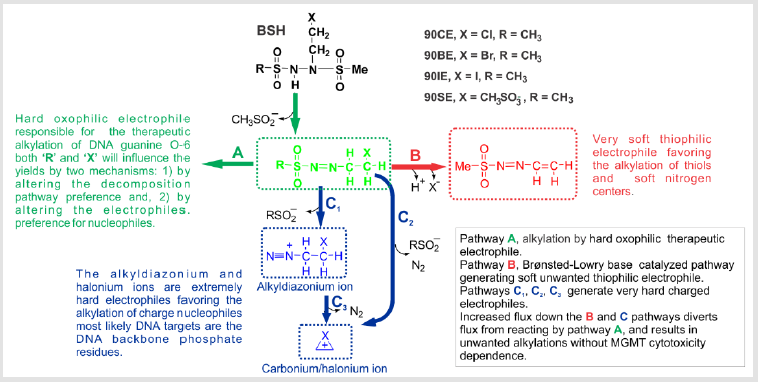
Figure 1: Major decomposition routes open to haloethyl BSHs and their functioning analogs.
Early decomposition studies on BSHs were conducted in weak Tris-HCl (pH 7.4) buffers where the newly identified Brønsted- Lowry base-catalyzed decomposition pathway was almost absent and therefore overlooked [54]. Recent studies in buffers with high Brønsted-Lowry base activity (such as phosphate) indicated that haloethyl BSHs (and some analogs thereof) decomposed almost entirely by an alternative pathway that is a relatively soft thiophilic, and aromatic nitrogen preferring electrophile (Figure 1 pathway B) in this environment [34]. This pathway does not generate the therapeutic oxophilic haloethylating species favoring the alkylation of DNA guanine O-6, but rather a reactive, softer electrophile that reacts via ‘Michael type’ addition reactions and most likely contributes to non-specific toxicities that show no MGMT activity dependence [34]. Even though the molar yield of guanine O-6 chloroethylations by 90CE and eventual yields of cross-links are ~ 2-fold greater than those obtained from BCNU they still represent a small proportion of the total overall alkylations [23,29]. However, the 90CE guanine O-6 alkylations account for a much greater proportion of its cytotoxicity as evidenced by the ~ 20-fold increase in resistance produced by MGMT expression, compared to only a 2- to 3-fold for the less guanine O-6 specific BCNU [13,30,31].
Eliminating or greatly reducing the non-specific alkylations/toxicities of 2-Chloroethylating BSHs would significantly raise the selectivity ratio for 90CE analogs beyond 20-fold. An increase in this ratio translates to significantly lower toxicity to normal MGMT-containing cells, increased potency, and a larger ‘toxicity threshold’ for targeted analogs. An increase in potency of 90CE analogs also facilitates formulation, and the targeting of effective quantities of cytotoxic material (targeting systems frequently have transportation limits) and also decreases non-specific toxicities from other non-therapeutic prodrug derived metabolites. KS90 is the methylating analog of 90CE and the primary oxophilic alkylating species generated is more restricted in its decomposition options. It cannot undergo the Brønsted-Lowry base-catalyzed halide loss, thus preventing pathway B from occurring, and while the formation of a methyldiazonium ion (CH3N+≡N) can occur, the high instability of the methyl carbonium ion precludes the formation of this extremely hard electrophilic species. Thus, in HL60 cells, KS90 generates a 5.5-fold greater molar yield of guanine O-6 alkylations than that produced by 90CE [13]. While KS90 and 90CE are not perfectly comparable since their primary oxophilic alkylating species (CH3SO2N=NCH3 and CH3SO2N=NCH2CH2Cl, respectively) likely differ somewhat in their hardness and preference for the O-6 position of DNA guanine, it does indicate that there is a significant potential for improvement in guanine O-6 alkylation yield of the chloroethylating analog.
Estimates of the losses due to pathway B can be readily made in model systems since pathways A and C(s) generate a readily detected haloalcohol on the alkylation of water whereas B results in halide liberation and a decrease in haloalcohol yield. In addition, the softer electrophile favoring the attack of thiols and aromatic nitrogens resulting from pathway B enables its assay either by thiol or 4-(4-nitrobenzyl)pyridine (NBP) trapping and HPLC or spectroscopic detection by the bleaching of chromophoric thiolates, or the generation of chromophoric NBP adducts [34]. The contribution of the known concentrations of inorganic phosphate and simple phosphoesters alone are likely to divert ~ 25% of the reaction flux down pathway B in the case of 90CE [34]. However, cells contain a vast number of other molecules that can act as Brønsted-Lowry bases and so the total contribution of pathway B would be expected to be significantly greater than ~25%. Direct experiments with intact cells are more complex, as an analysis of the reaction products does not take into account the proportions of alkylations occurring in the simple extracellular versus the complex intracellular, Brønsted-Lowry base-laden environments. However, we can make some approximations to obtain a range for losses by pathway B in cells based on other experimental observations. For example, the cross-linking of DNA in intact L1210 leukemia cells by 90CE occurs at ~ 1/8th the level as that of DNA in free solution under equivalent conditions in the absence of significant Brønsted-Lowry base activity [23].
This ~7/8th loss would set an upper limit for losses by pathway B in L1210 cells because other factors such as the shielding of DNA by associated proteins are also likely to come into play. Two 90CE analogs with very different sensitivities to Brønsted-Lowry base-catalyzed flux down pathway B (90SE and 90BE, with 50% maximal pathway B fluxes occurring at ~ 7.5 and ~ 37 mM phosphate, respectively) can also provide some clues as to the in vivo importance of pathway B (Figure 1). These analogs, 90BE and 90SE, possess almost identical cytotoxicity to L1210 cells, yet in the absence of significant Brønsted-Lowry base activity, 90SE cross-links DNA with ~ 4x the efficiency of 90BE. If we assume that the near identical biological activity of 90BE and 90SE results from them producing nearly equivalent levels of cross-links in cells, and that the ~ 4-fold greater cross-link yield of 90SE is negated by its greater sensitivity to Brønsted-Lowry bases diverting flux down pathway B, we can produce an estimate for the lowest level of the total Brønsted-Lowry base in L1210 cells in terms of its equiactive phosphate concentration. This value is approximately 50 mM phosphate-equivalent and would result in about 2/3rd of 90CE reacting via the undesired pathway B. Therefore, the elimination of or reduction of this pathway could have significant benefits (up to 3-fold).
The relative proportions of material reacting via pathways A and C are likely relatively fixed for a particular agent under physiological conditions and are not directly influenced by the presence of Brønsted-Lowry bases or other solutes. However, the structure of the primary haloethylating species RSO2N=NCH2CH2X will not only influence its preference for alkylating DNA guanine O-6, but also alter its tendency to fragment and react via the C pathways to generate very hard alkylating species which favor the alkylation of DNA phosphates. The alkylation of DNA phosphate follows the rank order 90IE > 90BE > 90CE (as expected, based on halonium ion stability favoring pathways C2 and C3) and an inverse relationship is seen in DNA cross-linking with 90CE > 90BE > 90IE. Even in the case of 90CE, DNA phosphate alkylations vastly outnumber DNA guanine O-6 alkylations, implying that at least theoretically, the cross-linking efficiency of RSO2N=NCH2CH2X can be greatly improved with appropriate substitutions. The fluxes down the three decomposition routes A, B and the C pathways can all easily be independently assayed/determined, allowing the identification of analogs that strongly favor the desired A pathway over the B/C pathways. In addition, the relative rates of the different guanine O-6 adducts generated by pathway A to progress to cross-links or be repaired by MGMT can also readily be evaluated [34,47]. Guanine O-6 adducts with the greatest MGMT ‘interceptability’ are expected to exhibit a greater inverse MGMT level dependent cytotoxicity. Thus, by maximizing the flux down pathway A, and the resultant lesion’s MGMT repairability, warheads/agents with an extreme inverse MGMT level dependent cytotoxicity will be produced, thereby minimizing toxicity towards normal MGMT-expressing cells and greatly enhancing toxicity to cancers selected for low or no MGMT expression.
Optimization of Pathway A
The therapeutic oxophilic electrophile generated by 90CE (CH3SO2N=NCH2CH2Cl) favors the chloroethylation of hard oxygen-based nucleophiles. Thus, water, in part because of its presence in a vast molar excess, intercepts the majority of these electrophiles, decreasing the yields of O6-(2-chloroethyl)guanine [23,34]. By tuning the electrophilic hardness of the generated electrophile to more strongly favor the alkylation of guanine O-6 relative to the alkylation of water, potency can be increased, and unwanted alkylations minimized.
Minimizing Unwanted Pathway B
We have compared the Brønsted-Lowry base (inorganic phosphate in these experiments) catalyzed flux down pathway B for 90CE and a number of analogs; 90BE, 90IE, and 90SE (Figure 2). It can be seen that the acidity of the proton beta to the halide (or methylsulfonate) rather than the leaving group ability of the halide or methylsulfonate controls the propensity of the molecule to react via the Brønsted-Lowry base catalyzed pathway B. In the case of 90IE (iodide is an exceptionally good leaving group), and an additional mechanism independent of that catalyzed by Brønsted-Lowry base(s) appears to be operative as evidenced by the observation that ~40% of the reaction proceeds via a soft electrophile generating pathway in the absence of a significant concentration of Brønsted- Lowry bases. Nonetheless, the influence of the Brønsted-Lowry base concentration on the increase in the flux down pathway B is still lower with 90IE than with the other analogs. These findings indicate that the rate-determining abstraction of the beta hydrogen by the Brønsted-Lowry base controls the rate of this reaction. Since this reaction competes with pathways A and C, the slowing of B will result in an increase in fluxes down the A and C reaction pathways and a reduction in flux via pathway B. There are three approaches (that can be used in combination) to slow pathway B.
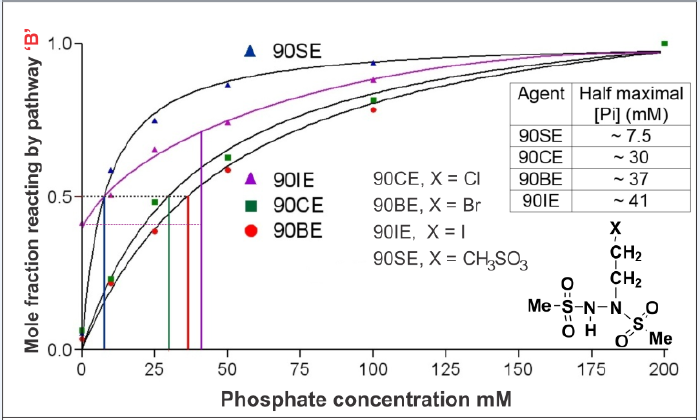
Figure 2: The effects of Phosphate acting as a Bronsted-Lowry base on the mole fraction of 90CE analogs that decomposes via reaction pathway B to generate a thiophilic electrophile. The inset table indicates the concentration pf phosphate that results in a half maximal phosphate-induce pathway switch. Note 90IE is somewhat unusual in that a significant proportion generates a thiophilic electrophile independent of the phosphate concentration. This likely represents a mechanism switch when “X” is a superb leaving group.
A. Decrease the acidity of the beta hydrogen in the primary
chloroethylating species by utilizing a pseudohalide, or other
leaving group, comparable in leaving group ability to Cl but less
electron-withdrawing.
B. Decrease the acidity of the beta hydrogen by replacing
the N-2 methylsulfonyl moiety with a less electron-withdrawing
moiety, e.g., a dimethylsulfamoyl moiety.
C. Replace the beta hydrogens (or all hydrogens on the
chloroethyl moiety) with deuteriums. Since the C-D bond is
significantly stronger than the C-H bond (~ 12 KJ/mol), the
activation energy required to break the former is larger, resulting in
a smaller rate constant [54,55].
Thus, a pronounced kinetic isotope effect is observed on
deuterium/hydrogen substitution when the breaking of this bond
is involved in the rate-determining step. The extent of reaction
slowing is quite variable and is likely dependent upon the degree of
breakage of the C-D bond in the transition state. Although reaction
rate differentials of < ~ 7-fold are usually observed, values as high
as 70-fold have been documented [56]. It is expected that by using
these methods, or a combination thereof, toxicities arising from,
and potency losses due to, flux down pathway B can be rendered
inconsequential.
Minimizing Unwanted Flux Down the C Pathways
Further fragmentation of the therapeutic alkylating species, CH3SO2N=NCH2CH2Cl, via the C pathways to generate very hard, charged electrophiles is influenced by the stability of the resultant diazonium or carbocations and the leaving group ability of the RSO2¯ anion. These structural changes are also likely to influence the softness/hardness of the resultant electrophile and hence its specificity for the DNA guanine O-6 position. Thus, the DNA guanine O-6 adduct yields will be influenced in two ways: the loss of the therapeutic alkylating species via fragmentation and its specificity for guanine O-6. With 90CE, as with the CNUs, pathways that generate extremely hard alkylating species strongly dominate over those generating guanine O-6 lesions in DNA in terms of number (> 100-fold) [10-12]. While the DNA backbone phosphate alkylations are of relatively low toxicity on a per lesion basis [57], they still are contributors to MGMT-independent toxicity, and represent a considerable loss in potential therapeutic efficacy and therefore should be minimized.
Optimization of BSH pKa Values
Weak acids like BSHs have advantages in treating solid tumors since these cells possess atypically large and inverted transplasmalemma pH gradients [58]. Normal tissues have an intracellular pH value of ~ 7.2 and an extracellular pH of ~ 7.5, while tumor tissues have intracellular pH values ~ 7.5 and extracellular pH values around 6.9. Weakly acidic agents become more protonated in the acidic extracellular environment, and thus more permeable, but upon entering the cell become more ionized and more strongly retained. These beneficial pH gradient effects are expected to be further exaggerated by the effects of pH on BSH half-life [47]. In the acidic extracellular tumor environment BSHs will be stabilized but upon cancer cell entry, their half-life will shorten. This will prevent the attainment of a pH biased concentration equilibrium and will maintain a concentration gradient to drive BSH entry [47]. The opposite effect should occur in normal cells although to a reduced extent. The pKa values of BSHs are easily manipulated by replacing the methyl groups of the N-1 and N-2 methylsulfonyl moieties with groups that are either more or less electron withdrawing. There are three major variables that determine the optimum pKa value for a BSH warhead;
To generate near maximal differences in pH biased transmembrane equilibrium concentration between normal and tumor cells (max ratio possible ~ 8-fold) the calculated pKa value should be less than ~0;
To ensure permeability the pKa value should not be too far below the extracellular pH of tumor tissue (6.9) since less BSH would then be in the permeable unionized form possibly giving insufficient time for equilibrium to be approached;
To maximize differential toxicity solely based on the differentials in intracellular half-life (max differential ~8-fold) between normal and tumor cells the best pKa value is 7.35.
The relative importance and interplay of these three major factors is complex. Therefore, experimentation would be required determine the optimal BSH pKa range using analogs in in vitro clonal survival assays conducted over relevant extracellular pH values. Preliminary experiments utilizing 90CE (pKa ~ 6.7) revealed extracellular pH dependent changes in cytotoxicity consistent with the expected pH biased transplasmalemma equilibria [47]. It should be noted that BSH half-life has a greater dependency upon the leaving group ability than the electron withdrawing effect of the N-1 substituent, while the pKa shows the converse relationship. Therefore, these two important characteristics do not have to move in parallel. Weakly basic compounds exhibit the opposite accumulation effect to those of weak acids like the BSHs, in response to pH gradients, and this attenuates their cancer cell selectivity. Thus, anticancer drugs such as doxorubicin and vinblastine, are negatively impacted, reducing their desirability as tumor targeted warheads [58].While the deuteration of the chloroethyl group is expected to primarily influence only decomposition pathway B directly, other changes will more strongly influence multiple facets of the decomposition pathways to various extents [59]. The use of computational chemistry is expected to aid not only in the selection of compounds for synthesis by predicting effects that a purely intuitive approach to compound selection may miss, but also in the interpretation of structure-activity relationships [60]. It is likely that the reaction energy barrier to cyclization following the alkylation of the O-6 position of guanine in deuterated 90CE analogs will also be raised. Slowing this reaction is expected to increase the MGMT temporal repair window, likely reducing toxicity to MGMT expressing cells.
The Structural Modifications to the BSH Moiety Meriting Further Investigation are listed below
Replacement of the halogen with a pseudohalogen or other leaving group in order to study how a shift in charge density and electrophilicity affects the alkylation of the O-6 position of DNA guanine relative to the unwanted alkylation of water (and further fragmentation to very hard electrophilic species).
The effects of modification of the ethylene moiety (attached to the halogen) on the electrophilicity of the pathway A reactive species and its ability to alkylate guanine O-6. This modification is also likely to alter the rate of the cyclization reaction on guanine following alkylation and the MGMT repair rate.
Replacement of the terminal methyl group on the N2-methylsulfonyl moiety of 90CE with a more electron-releasing group, with a view to reducing the leaving group ability of this moiety in the resultant chloroethylating species (ClCH2CH2N=NSO2R, where R = a group more electron-releasing than CH3) and thus reduce the formation of unwanted, very hard chloroethylating species leading to the alkylation of DNA backbone phosphates.
Replacement of the chloroethyl hydrogens by deuterium to study how this change affects the rate of the base-catalyzed HCl elimination that generates unwanted thiophilic and aromatic nitrogen preferring alkylating species.
Investigation of how the fluxes down the different decomposition pathways are altered following the above modifications.
Calculation of pKa values to guide choice of compounds for synthesis for pka optimization studies.
Solubility and partition coefficient calculations of simple prodrug derivatives of the novel BSHs to identify agents possessing molecular properties important for good pharmacokinetics.
Experimental Approaches
The experimental approaches to investigate the decomposition
pathway fluxes and alkylation preferences of the newly synthesized
90CE analogs are listed below.
A. Synthesis of deuterated 90CE and initial agents of interest
arising from computational studies.
B. Analysis of agent decomposition products (thiol-trapped
adducts) under various Brønsted-Lowry base concentration
([Pi]) conditions to determine structure-activity effects on the
flux down pathway B.
C. Measurement and analysis of salient DNA lesion products (G-C
cross-links and phosphate alkylation products) under various
Brønsted-Lowry base concentration conditions to determine
structure-activity effects on the flux down pathways A and
C1-3 and the relative molar fluxes between different analogs. The correlation of experimental data concerning nucleophile
target preference with computational chemistry calculations
of alkylating intermediate electrophilicity will greatly aid in
the choice of future 90CE analogs for synthesis and testing.
D. Measurement of the guanine O-6 lesion repair rate by MGMT
(and MGMT variants) and the kinetics of the progression of
the primary lesion to G-C cross-links in simple cell free in vitro
model systems. In kinetic competition assays (between MGMT
repair and progression to cross-links), primary lesions which
are most readily intercepted by MGMT are expected to exhibit
a greater inverse MGMT activity to cytotoxicity dependency.
E. Measurement of the differential toxicities of 90CE analogs (and
simple prodrugs thereof) to a large panel of matched cell lines
with and without various levels of MGMT expression.
F. Testing of simple laromustine-like prodrugs of the most
promising 90CE analogs in murine models with tumors
expressing and not expressing MGMT.
G. Measurement of solubility and octanol/water partition
coefficient of simple prodrug forms of novel BSHs.
H. Measurement of BSH half-life and pKa values (determined
from the pH dependency of decay rate).
Chemical Synthesis
No complex chemistry is expected to be required for the synthesis of the described BSHs, and a minimally equipped chemical synthesis laboratory should suffice [35,47,48,54,60,61]. A new synthetic route to 90CE using readily available di-tert-butylhydrazine-1,2- dicarboxylate as the starting material is illustrated in Figure 3, this should allow the facile synthesis of the deuterated analogs, using readily available deuterated 2-bromoethanol. This method is based upon a modification of a published high yield method [62,63] (Figure 3).
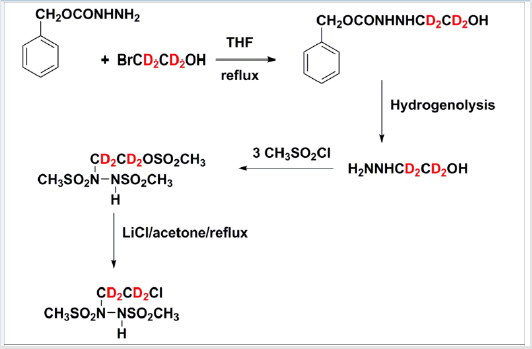
Figure 3: Proposed Scheme for the Synthesis of deuterated 90CE using readily available (2-bromo)(1,1,2,2-tetra-deutero)ethanol as a starting material .The overall yield in test syntheses using non-deuterated 2-bromoethanol is ~25%.
References
- Gerson SL (2002) Clinical relevance of MGMT in the treatment of cancer. J Clin Oncol 20: 2388-2399.
- Shyam K, Penketh PG, Baumann RP, Finch RA, Zhu R, et al. (2015) Antitumor Sulfonylhydrazines: Design, Structure-Activity Relationships, Resistance Mechanisms, and Strategies for Improving Therapeutic Utility. J Med Chem J Med Chem 58: 3639-3671.
- Ishiguro K, Shyam K, Penketh PG, Sartorelli AC (2008) Development of an O6-alkylguanine-DNA alkyltransferase assay based on covalent transfer of the benzyl moiety from [benzene-3H] O6-benzylguanine to the protein. Anal Biochem 383: 44-51.
- Christmann M, Verbeek B, Roos WP, Kaina B (2011) O6-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: enzyme activity, promoter methylation and immunohistochemistry. Biochim Biophys Acta 1816: 179-180.
- Penketh P, Williamson H, Baumann RP, Shyam K (2021) Design Strategy for the EPR Tumor-Targeting of 1,2-Bis(sulfonyl)-1-alkylhydrazines. Molecules 26(2): 259-275.
- Chan CL, Wu Z, Eastman A, Bresnick E (1992) Irradiation-induced expression of O6-methyltransferase in mammalian cells. Cancer Res 52: 1804-1809.
- Eisenstein M (2014) Clinical trials: more trials, fewer tribulations. Nature 509: S55-S57.
- Weinkam RJ, Lin HS (1979) Reactions of 1,3-bis(2-chloroethyl)-1-nitrosourea and 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea in aqueous solution. J Med Chem 22: 1193-1198.
- Brundrett RB, Cowers JW, Colvin M, Jardin I (1976) Chemistry of nitrosoureas, Decomposition of deuterated 1,3-bis(2-chloroethyl)-1-nitrosoureas. J Med Chem 19: 958-964.
- Ludlum DB (1997) The chloroethylnitrosoureas: sensitivity and resistance to cancer chemotherapy at the molecular level. Cancer Invest 15: 588-598.
- Weinkam RJ, Lin H (1982) Chloroethylnitrosourea cancer chemotherapeutic agents. Adv Pharm Chemother 19: 1-33.
- Bodell WJ, Tokuda K, Ludlum DB (1988) Differences in DNA alkylation products formed in sensitive and resistant human glioma cells treated with N-(2-chloroethyl)-N-nitrosourea. Cancer Res 48: 4489-4492.
- Ishiguro K, Zhu YL, Shyam K, Penketh PG, Baumann RP, et al. (2010) Quantitative relationship between guanine O6-alkyl lesions produced by Onrigin™ and tumor resistance by O6-alkylguanine-DNA alkyltransferase. Biochem Pharmaco 80: 1317-1325.
- Penketh P, Williamson H, Shyam K (2020) Physicochemical considerations of tumor selective drug delivery and activity confinement with particular reference to 1,2-bis(sulfonyl)-1-alkylhydrazines delivery. Curr. Drug Deliv 17(5): 362-374.
- Wright J (2014) Nanotechnology: Deliver on a promise. Nature 509: S58-S59.
- Fang J, Nakamura H, Maeda H (2011) The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 18: 136-151.
- DAtri S, Tentor L, Lacal PM, Graziani G, Pagani E, et al. (1998) Involvement of the mismatch repair system in temozolomide-induced apoptosis. Mol Pharmacol 54: 334-341.
- Toorchen D, Lindamood C, Swenberg JA, Topal MD (1984) O6-Methylguanine-DNA transmethylase converts O6-methylguanine thymine base pairs to guanine thymine base pairs in DNA. Carcinogenesis 5: 1733-1735.
- Branch P, Aquilina G, Bignami M, Karran P (1993) Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature 362: 652-654.
- Kolodner R (1996) Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev 10: 1433-1442.
- Yan T, Berry SE, Desai AB, Kinsella TJ (2003) DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin Cancer Res 9: 2327-2334.
- Barvaux VA, Ranson M, Brown R, Mc Elhinney RS, Mc Murry TB, et al. (2004) Dual repair modulation reverses Temozolomide resistance in vitro. Mol Cancer Ther 3: 123-127.
- Penketh PG, Baumann RP, Ishiguro K, Shyam K, Seow HA, et al. (2008) Lethality to leukemia cell lines of DNA interstrand cross-links generated by Cloretazine derived alkylating species. Leukemia Res 32: 1546-1553.
- Ludlum DB (1997) The chloroethylnitrosoureas: sensitivity and resistance to cancer chemotherapy at the molecular level. Cancer Invest 15: 588-598.
- Noll DM, Mason TM, Miller PS (2006) Formation and repair of interstrand cross-links in DNA. Chem Rev 06: 277-301.
- Gonzaga PE, Brent TP (1989) Affinity purification and characterization of human O6-alkylguanine-DNA alkyltransferase complexed with BCNU-treated, synthetic oligonucleotide. Nucleic Acids Res 17: 6581-6590.
- Rockwell S, Liu Y, Seow H A, Ishiguro K, Baumann RP, et al. (2012) Preclinical evaluation of Laromustine for use in combination with radiation therapy in the treatment of solid tumors. Int J Radiat Biol 88: 277-285.
- Olivier JC (2005) Drug Transport to Brain with Targeted Nanoparticles. NeuroRx Jan 2: 108-119.
- Penketh PG, Shyam K, Sartorelli AC (2000) Comparison of DNA lesions produced by tumor-inhibitory 1,2-bis(sulfonyl)hydrazines and chloroethylnitrosoureas. Biochem Pharmacol 59: 283-291.
- Naghipur A, Ikonomou MG, Kebarle P, Lown JW (1990) Mechanism of action of (2-haloethyl)nitrosoureas on DNA: discrimination between alternative pathways of DNA base modification by 1,3-bis(2-fluoroethyl)-1-nitrosourea by using specific deuterium labeling and identification of reaction products by HPLC/tandem mass spectrometry. J Am Chem Soc 112: 3178-3187.
- Ishiguro K, Shyam K, Penketh PG, Sartorelli AC (2005) Role of O6-alkylguanine-DNA alkyltransferase in the cytotoxic activity of cloretazine. Mol Cancer Ther 4:1755- 1763.
- Ishiguro K, Seow HA, Penketh PG, Shyam K, Sartorelli AC (2006) Mode of action of the chloroethylating and carbamoylating moieties of the prodrug cloretazine. Mol Cancer Ther 5: 969-976.
- Glassner BJ, Weeda G, Allan JM, Broekhof JL, Carls NH, et al. (1999) DNA repair methyltransferase (Mgmt) knockout mice are sensitive to the lethal effects of chemotherapeutic alkylating agents. Mutagenesis 14: 339-347.
- Penketh PG, Shyam K, Zhu R, Baumann RP, Ishiguro K, et al. (2014) Influence of phosphate and phosphoesters on the decomposition pathway of 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazine (90CE), the active anticancer moiety generated by laromustine, KS119 and KS119W. Chem Res Toxicol 27: 818-833.
- Shyam K, Penketh PG, Loomis RH, Rose WC Sartorelli AC (1996) Antitumor 2-Aminocarbonyl-1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazines. J Med Chem 39: 796-801.
- Finch RA, Shyam K, Penketh PG, Sartorelli AC (2001) 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-(methylamino)carbonylhydrazine (101M): A Novel Sulfonylhydrazine Prodrug with Broad-Spectrum Antineoplastic Activity. Cancer Res 61: 3033-3038.
- Baril BB, Baril EF, Laszlo J, Wheeler GP (1975) Inhibition of rat liver DNA polymerase by nitrosoureas and isocyanates. Cancer Res 35: 1-5.
- Kann HE Jr, Kohn KW, Lyles JM (1974) Inhibition of DNA repair by 1,3-bis(2-chloroethyl)-1-nitrosourea breakdown product, 2-chloroethyl isocyanate. Cancer Res 34: 398-402.
- Kann HE Jr, Kohn KW, Widerlite L, Gullion D (1974) Effects of 1,3-bis(2-chloroethyl)-1-nitrosourea and related compounds on nuclear RNA metabolism. Cancer Res 34: 1982-1988.
- Schallreuter KU, Gleason FK, Wood JM (1990) The mechanism of action of the nitrosourea anti-tumor drugs on thioredoxin reductase, glutathione reductase and ribonucleotide reductase. Biochim Biophys Act 1054: 14-20.
- Gibson NW, Hickman JA (1982) The role of isocyanates in the toxicity of antitumor haloalkylnitrosoureas. Biochem. Pharmacol 31: 2795-2800.
- Johnston TP, Montgomery JA (1986) Relationship of structure to anticancer activity and toxicity of the nitrosoureas in animal systems. Cancer Treat Rep 70: 13-31.
- Giles F, Verstovsek S, Thomas D, Gerson S, Cortes J, et al. Phase I study of cloretazine (VNP40101M), a novel sulfonylhydrazine alkylating agent, combined with cyarabine in patients with refractory leukemia. Clin Cancer Res 11: 7817-7824.
- Schiller GJ, O Brien SM, Pigneux A, Deangelo DJ, Vey N, et al. (2010) Single-agent laromustine, a novel alkylating agent, has significant activity in older patients with previously untreated poor-risk acute myeloid leukemia. J Clin Oncol 28: 815-821.
- Seow H A, Penketh PG, Shyam KS, Rockwell S, Sartorelli AC (2005)1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4-nitrophenyl)ethoxy]carbonyl]hydrazine (KS119): A Novel Antineoplastic Agent with Selective Cytotoxicity to Hypoxic Cells. Proc Natl Acad Sci USA 102(26): 9282-9287.
- Kim EY, Liu Y, Akintujoye OM, Shyam K, Grove TA, et al. (2012) Preliminary studies with a new hypoxia-selective cytotoxin, KS119W, in vitro and in vivo. Radiation Res 178: 126-137.
- Penketh PG, Finch RA, Sauro R, Baumann RP, Ratner ES, et al. (2018) pH-dependent general base catalyzed activation rather than isocyanate liberation may explain the superior anticancer efficacy of laromustine compared to related 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazine prodrugs. Chem Biol Drug Des 91(1): 62-74.
- Shyam K, Penketh PG, Divo AA, Loomis RH, Patton CL, et al. (1990) Synthesis and evaluation of 1,2,2-tris(sulfonyl)hydrazines as antineoplastic and trypanocidal agents. J Med Chem 33: 2259-2264.
- Coles B (1985) Effects of modifying structure on electrophilic reactions with nucleophiles. Drug Metab Rev 15: 1307-1334.
- Shyam K, Penketh PG, Loomis RH, Sartorelli AC (1998) Thiolysable prodrugs of 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazine with antineoplastic activity. Eur J Med Chem 33: 609-615.
- Shyam K, Penketh PG, Divo AA, Loomis RH, Rose WC, et al. (1993) Synthesis and evaluation of 1-acyl-1,2-bis(methylsulfonyl)-2-(2-chloroethyl)hydrazines as antineoplastic agents. J Med Chem 36: 3496-3502.
- Baumann RP, Ishiguro K, Penketh PG, Shyam K, Zhu R, et al. (2011) KS900: a hypoxia-directed, reductively activated methylating antitumor prodrug that selectively ablates O6-alkylguanine-DNA alkyltransferase in neoplastic cells. Biochem Pharmacol 81: 1201-1210.
- Baumann RP, Penketh PG, Ishiguro K, Shyam K, Zhu YL, et al. (2010). Reductive activation of the prodrug 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4-nitrophenyl)ethoxy]carbonyl]-hydrazine (KS119) selectively occurs in oxygen-deficient cells and overcomes O6-alkylguanine-DNA alkyltransferase mediated KS119 tumor cell resistance. Biochem Pharmacol 79: 1553-1561.
- Penketh PG, Shyam K, Sartorelli AC (1994) Studies on the Mechanism of Decomposition and Structural Factors Affecting the Aqueous Stability of 1,2-Bis(sulfonyl)-1-alkylhydrazines. J Med Chem 37: 2912-2917.
- Penketh PG, Patridge E, Shyam K, Baumann RP, Zhu R, et al. (2014) Influence of glutathione and glutathione S-transferases on DNA interstrand cross-link formation by 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazine, the active anticancer moiety generated by laromustine. Chem. Res. Toxicol 27: 1440-1449.
- Gant TG (2014) Using Deuterium in Drug Discovery: Leaving the Label in the Drug. J Med Chem 57: 3595-3611.
- Jones GD, Le Pla RC, Farmer PB (2010) Phosphotriester adducts (PTEs): DNA's overlooked lesion. Mutagenesis 25: 3-16.
- Webb BA, Chimenti M, Jacobson MP, Barber DL (2011) Dysregulated pH: a perfect storm for cancer progression. Nature Rev Cancer 11: 671-677.
- Patridge EV, Eriksson ES, Penketh PG, Baumann RP, Rui Zhu, et al. (2012) 7-Nitro-4-(phenylthio)benzofurazan is a potent generator of superoxide and hydrogen peroxide. Arch Toxicol 86: 1613-1625.
- Penketh PG, Baumann RP, Shyam K, Williamson HS, Ishiguro K, et al. (2011) 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4-nitrophenyl)ethoxy]carbonyl]-hydrazine (KS119): a cytotoxic prodrug with two stable conformations differing in biological and physical properties. Chem Biol Drug Des 78: 513-526.
- Shyam K, Penketh PG, Shapiro M, Belcourt MF, Loomis RH, et al. (1999) Hypoxia-selective nitrobenzyloxycarbonyl derivatives of 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazine. J Med Chem 42: 941-946.
- Rasmussen LK (2006) Facile synthesis of mono-, di-, and trisubstituted alpha-unbranched hydrazines. J Org Chem 71: 3628-3629.
- Bredihhin A, Mäeorg U (2008) Effective strategy for the systematic synthesis of hydrazine derivatives. Tetrahedron 64: 6788-6793.
