 Research Article
Research ArticleAbstract
Aim: Congenital diarrheal disorders (CDDs) represent a group of enteropathies
caused by different pathomechanisms that typically have their onset in the neonatal
period, and that carry significant morbidity and mortality rates. We aimed to evaluate
the clinical, prognostic and molecular features of patients who presented with chronic
diarrhea and were diagnosed with CDDs as a result of genetic analysis.
Methods: The study comprized family and nutritional history, detailed clinical
history, and physical examination findings in patients who presented with intractable
or episodic diarrhea. Routine laboratory tests, detailed stool tests, gastroscopy and
colonoscopy were performed for all cases.
Results: As a result of genetic testing; 1 patient was diagnosed with DGAT-1 enzyme
deficiency, 1 patient with microvillus inclusion disease (MVID), 1 patient with trichohepato-
enteric (THE) syndrom type 2, 1 patient cystic fibrosis, 2 patients with enteric
anendocrinosis (EA), 1 patient with congenital tufting enteropathy, 1 with patient
Mednik syndrome and 1 patient was diagnosed with glucose-galactose malabsorption.
Four of the cases died during the follow-up period.
Conclusions: In cases presenting with chronic diarrhea, awareness of the CDDs
should be increased and molecular analysis should be included in the diagnostic
algorithm after carefully taken detailed history and routine examination. Thus, it will
contribute to the reduction of morbidity and mortality rates by enabling CDD-specific
treatment.
Keywords: Chronic Diarrhea; Congenital Diarrheal Disorders (CDDs); Congenital Diarrhea; Mutations; Molecular Diagnosis
Introduction
Diarrheal disorders are the second most common leading cause of death worldwide with an estimated 750,000 deaths annually in children under 5 years old. Diarrhea is defined as stool volume >10 g/kg/day in infants, and >200 g/kg /day in adolescents [1,2]. The terms “persistent” and “chronic”are often used interchangeably, but according to the World Health Organization (WHO) persistent diarrhea persists for more than two weeks and chronic diarrhea usually exceeds 1 month (> 3 times / day watery) without acute onset [2]. In children chronic diarrhea can occur due to large number of disorders including infections, postinfection syndrome, diet related factors (allergy or intolerance), inflammatory bowel disease, malabsorption, cystic fibrosis, immune deficiencies and congenital diarrheal disorders (CDDs) [3]. Moreover, numerous etiologies, overlapping symptoms, and similar clinical presentations make the diagnosis and management of the disease difficult. Therefore, detailed medical history and physical examination findings are crucial in the evaluation of cases [4]. CDDs are rare, mostly autosomal recessive inherited enteropathies that typically early onset with varying severity and prognostic features [5]. In the etiology of CDDs which are associated with high risk of mortality and morbidity; Over 30 CDD disordes and 35 responsible genes have been identified that cause defects in nutrients and electrolytes digestion, absorption, enterocyte structures, intestinal immunerelated homeostasis and enteroendocrine cell development [6]. Thus, molecular based diagnosis is required and clearly plays a key role in the CDD diagnosis. Therefore, we aimed to evaluate the clinical, prognostic and molecular features of patients who were presented with chronic diarrhea and diagnosed with CDDs as a result of genetic analysis [7].
Materials and Methods
This study was performed with the Institutional Review Board
protocol approval date 18/02/2019 and number 2019/13 in
Istanbul Dr. Sadi Konuk Training and Research Hospital, Department
of Pediatric Gastroenterology, Hepatology and Nutrition, between
01/06/2017 to 01/06/2019. A total number of 9 patients (5 girl
- 4 boy), who were presented with dehydration, severe diarrhea,
weight loss for more than 4 weeks and diagnosed with CDDs,
evaluated retrospectively. During the application, detailed clinical
history was taken for the primary diagnostic assessment in cases.
The information including demogrophic data, disease onset time,
nutritional history, consanguinity, family atopy, asthma, eczema
history, death of another brother or sister, presence of similar
disease in family, maternal history of polyhydroamniosis, history of
stool loose in the first 24 hours, mucus and blood content in gaita,
stool volume and frequency, vomiting, and recurrent lung infection
were questioned in interviews with the parents.
In the physical examination of the patients, abdominal
distention, perianal hyperemia, rash, pretibial edema, severe
gland dermatitis, skin dryness, signs of rickets, ADEK vitamin
deficiency, and other syndromic findings were evaluated. Detailed
biochemical analysis, complete blood count, ESR, and allergy panel
were analysed in all cases. Where necessary, celiac antibodies, skin
prick test and sweat test were examined. In addition to detailed
morphological and microscopic assessment in stool, bacteriological
evaluation was performed by gaita culture. Genetic examination
was performed in pateints who could not be diagnosed as a result
of detailed clinical history, routine laboratory tests, gastroscopy,
colonoscopy, and histopathological evaluation. Blood cell count
analysis was performed on patients’ venous blood samples.
Haematological parameters were analyzed using a hematology
analyzer (Cell-Dyne 3700, Abbott, Abbott Park, IL, USA).
Biochemical analysis performed from serum samples by electrochemiluminescence
immunoassay on Beckman Coulter Unicel DXI
800 analyzer. CDDs-related mutations were examined with the help
of real-time Polymerase Chain Reaction (PCR) technique through
genomic DNA isolated from the patients’ peripheral blood.
Results
The mean age of 9 CDD-patients (5 female and 4 male) included in our study was 19 months (Ranged = 1-84). The oral intake was stopped in 6 patients who applied with watery diarrhea and followed up for 24 hours. Three of the patients were diagnosed with osmotic diarrhea that stopped with fasting, and 3 patients were diagnosed with secretory diarrhea which continued with fasting. In addition, 3 patients applied with fatty stool diarrhea and diagnosed with osmotic diarrhea due to the high steatocrit (Table 1).
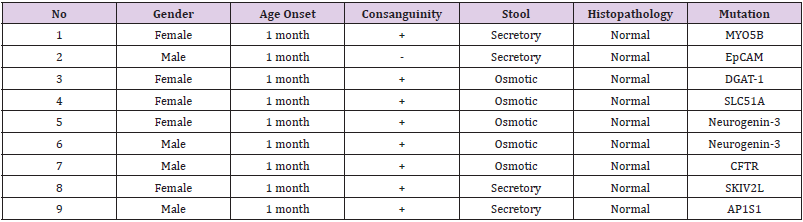
Table 1: CFTR Mutations identified in patients.
Case 1
First patient was presented with secretory diarrhea. Family history revealed consanguineous parents and death of MVIDdiagnosed brother. There was no oral intake and no response to formula feeding. Thus, parenteral nutrition (PN) was maintained. Genetic examination revealed a homozygous mutation in MYO5B gene and patient diagnosed with MVID. She died due to sepsis at the age of 5-months.
Case 2
Second patient was also presented with secretory diarrhea which continued with fasting and without response to formula feeding. He has a history of death brother due to the CDD-like disease but there was no consanguinity among the parents. The patient has poor anthropometric status. Additionally, histopathological findings in gastroscopy and colonoscopy were normal. Genetic analysis identified a homozygous mutation in the EPCAM gene and patient diagnosed with congenital tufting enteropathy. He continuous on TPN administration and oral table food, while awaiting bowel transplant.
Case 3
She was presented with peripheral edema and osmotic fatty diarrhea which stopped with fasting. Family history revealed consanguinity in parents. Gastroscopic and colonoscopic examination showed no abnormal finding. There was low plasma lipid level, lymphopenia, fecal alpha 1 antitrypsin, increased steatocrit and normal fecal elastase levels in routine examinations. Genetic analysis revealed a homozygous mutation in DGAT-1 gene and patient diagnosed with DGAT-1 enzyme deficiency. She still maintains her treatment with low-fat diet + high MCT and high protein table foods.
Case 4
A 7-years-old girl presented with distention and episodes of diarrhea which onset and maintained since the first days of her life. There was no response to regular formula regimens, which was started as a results of positive reducing substance in stool (Ph=4). Genetic analysis identified a homozygous SCL5A1 mutation, and she was diagnosed with glucose-galactose malabsorption. The patient is being followed up with administration of lactose-free milk, glucose-poor and fructose-based diet.
Case 5 and 6
Our 5th and 6th cases are brother and sister born to consanguineous parents. They were presented with watery diarrhea onset in early infancy which stopped with fasting. Gastroscopic and colonoscopic examination findings were normal. Molecular analysis revealed homozygous mutation in the Neurogenin-3 gene, and they were both diagnosed with EA. Since both of the patients are TPN dependent, their hospitalization continues.
Case 7
Our 7th case was presented with osmotic fatty diarrhea which stopped with fasting. During postnatal 4th month he was under the birth weight. Family history revealed consanguinity in parents. In routine examinations steatocrit was positive and fecal elastase was < 100 mg/g. Molecular genetic analysis has identified homozygous mutations in the CFTR gene and he was diagnosed with Cystic fibrosis. Although PERT (pancreatic enzyme replacement therapy), ADEK vitamin supplementation, pulmozym, pulmonary rehabilitation support and hypercaloric formula regimens was started, he died due to sepsis.
Case 8
Our 8th case was presented with hair anomalies, facial dysmorphism, hepatomegaly, cholestasis and failure to thrive. She was also born to consanguineous parents. Gastroscopic and colonoscopic examination showed no abnormal finding. Genetic examination revealed a homozygous mutation in SKIV2L gene and patient diagnosed with THE syndrom type 2. She died due to sepsis.
Case 9
Our last case presented with secretory diarrhea, ichthyosis, and weight loss. Family history revealed consanguinity in parents. There was no response to variety of formula regimens. Genetic analysis identified AP1S1 mutation, and he was diagnosed with Mednik syndrome. He received TPN administration but died due to sepsis.
Discussion
CDDs are severe and rare group of life-threatening enteropathy
with high mortality.5 Supportively Ye et al. screened 137 CDDschildren
with an average symptom onset age of 28 days; they
reported successful rate of molecular analyzes as 64.2% (88/137)
and the overall mortality rate was 14.6%. In recent years, an
increasing number of new genomes associated with CDDs have
been identified. In addition, according to the classification
suggested by Berni Canani published in 2010, CDDs are grouped
in 4 categories considering the type of defect; the first group
includes digestive-absorption defects and nutrient-electrolyte
transport defects. Glucosegalactose malabsorption (SLC5A1 gene)
and cystic fibrosis (CFTR gene) are defined in this group [8].
Glucose-galactose malabsorption is an extremely rare disorder
in enterositis, which occurs with sodium-coupled transport
defect of glucose and galactose. It is an autosomal recessive (OR)
inherited disorder caused by mutation in SLC5A1, which is the Na+ / glucose cotransporter gene. In a study, Anderson et al. revealed
no abnormal findings by routine laboratory, imaging, endoscopy
and histopathological evaluation in a patient at the age of 37-days
who presented with weight loss, hyperbilirubinemia, metabolic
acidosis and congenital diarrhea. However, in molecular analysis,
a homozygous SLC5A1 mutation has reported [9]. Similarly, in our
4th case; a 7-years-old girl presented with episodes of diarrhea
which onset in the first days of her life. Genetic analysis identified
a homozygous SCL5A1 mutation, and she was diagnosed with
glucose-galactose malabsorption. She is being followed up with
administration of lactose-free milk, glucose-poor and fructosebased
diet.
Disorders in CDD classification group 2 include differentiation
and polarization defects of enterocytes; MVID (MYO5B gene),
congenital tufting enteropathy (EpCAM gene) and syndromic
diarrhea are defined in this group [8]. MVID is a rare OR inherited
disease which can cause severe congenital diarrhea with high
morbidity and mortality. Sadiq et al. reported a 17-day-old, female
infant presented with severe diarrhea with unknown etiology,
metabolic derangements and failure to thrive; despite detailed
stool examination, abdominal usg, duodenal biopsy and flexible
sigmoidoscopy examinations, researchers failed to diagnose.
Even though researchers tried various treatment and formula
regimens, none of them was effective to stop diarrhea. Finally, by
applying molecular methods, homozygous MYO5B gene mutation
was detected in the patient, and she was diagnosed with MVID.
As the bowel transplantation is only treatment method reached
consensus for MVID; it was reported that patient was maintained
on TPN while awaiting bowel transplant [10]. Similarly in our
first case presented with secretory diarrhea; she had history of
consanguinity in parents and death of MVID-diagnosed brother.
Genetic examination revealed a homozygous mutation in MYO5B
gene and patient diagnosed with MVID. But she died due to sepsis
at the age of 5-months. Similarly, congenital tufting enteropathy
(CTE) formed by OR inherited EpCAM gene mutation; causes
severe diarrhea in the infant as a result of villous and epithelial
tufts changes [11]. Kahvecioğlu et al. reported no abnormal
findings on routine evaluation in a patient presented with watery
diarrhea (10 times a day) in postnatal 4th day. Researchers noted a
history of death brother and consanguinity in parents. They started
phosphorus supplementation on admission, but patient died due to
cardiac arrest on the postnatal 35th day. The patient was diagnosed
with CTE as a result of postmortem histopathological evaluations
[12]. Consistently in our study, our second case, who had a history
of death brother due to the CDD-like disease, was also presented
with secretory diarrhea. Genetic analysis identified a homozygous
mutation in the EPCAM gene, and he diagnosed with CTE. He
continuous on TPN administration and oral table food, while
awaiting bowel transplant.
Syndromic diarrhea or tricho-hepato-enteric (THE) syndrome
is caused by mutations in the SKIV2L and TTC37 genes. Patients
are presented with severe diarrhea in addition to the characteristic
phenotype [13]. Vardi et al. reported a 4-month-old girl with
congenital diarrhea, failure to thrive and history of consanguinity in
parents; Researchers confirmed SKIV2L gene mutation as a result
of molecular analysis [14]. In accordance with published data, our
8th case was presented with hair anomalies, facial dysmorphism,
hepatomegaly, cholestasis and failure to thrive. She was also
born to consanguineous parents. Genetic examination revealed
a homozygous mutation in SKIV2L gene. She died due to sepsis.
Disorders in CDD classification group 3 include differentiation and
function defects of enteroendocrine cells; enteric anendocrinosis
(EA) or congenital malabsorptive diarrhea (NEUROG3 gene)
are defined in this group. In addition, the last CDD classification
group includes dysregulation in intestinal immune responses [8]
OR inherited Neurogenin-3 (NEUROG3) gene mutation results in
EA characterized by severe malabsorptive diarrhea and intestinal
enteroendocrine cell deficiency. German-diaz et al. reported
Neurogenin-3 mutation in an 11-day-old boy presenting with
severe dehydration, metabolic acidosis and severe watery diarrhea.
At the end of the follow-up period, he was 16 years old and
reported to continue TPN once a day alongside gluten free regular
home diet [15]. Similarly, our 5th and 6th cases were presented
with watery diarrhea onset in early infancy. Molecular analysis
revealed homozygous mutation in the Neurogenin-3 gene, and they
were both diagnosed with EA. Since both of the patients are TPN
dependent, their hospitalization continues.
DGAT1 and DGAT2 (diacylglycerol acyltransferase) catalyze
triglyceride biosynthesis and OR mutations causing DGAT1
functional loss result in severe diarrhea and protein-losing
enteropathy [16]. Ratchford et al. identified a homozygous DGAT1
mutation in a 7-month-old girl presented with severe diarrhea.
TPN was started and changed to a low-fat diet of table foods by
13 months of age. At the end of the study, researchers reported
normal values of D vitamin, calcium, IgG, albumin and TG [17]. She
was presented with peripheral edema and osmotic fatty diarrhea
which stopped with fasting. Family history revealed consanguinity
in parents. Gastroscopic and colonoscopic examination showed no
abnormal finding. There was low plasma lipid level, lymphopenia,
fecal alpha 1 antitrypsin, increased steatocrit and normal fecal
elastase levels in routine examinations. Genetic analysis revealed a
homozygous mutation in DGAT-1 gene and patient diagnosed with
DGAT-1 enzyme deficiency. She still maintains her treatment with
low-fat diet + high MCT and high protein table foods. Consistently,
our 3th case was presented with osmotic fatty diarrhea which
stopped with fasting. Genetic analysis revealed a homozygous
mutation in DGAT-1 gene. She still maintains her treatment with
low-fat diet + high MCT and high protein table foods.
MEDNİK syndrome is caused by OR inherited mutation in the
AP1S1gene [18]. Incecik et al. reported a 10-year-old girl presented
with diarrhea, ichthyosis, and sensorineural deafness. Her family
history revealed consanguinity in parents and genetic analysis
identified a homozygous mutation in AP1S1gene. Researchers
started zinc sulphate during follow-up period [19]. Similarly, in
our last case presented with secretory diarrhea, ichthyosis, and
weight loss. Family history revealed consanguinity in parents.
Genetic analysis identified AP1S1 mutation. He received TPN
administration but died due to sepsis.
Conclusion
In conclusion, although chronic diarrheal diseases may present overlapping symptoms, CDDs are closely associated with poor prognosis and high mortality. Therefore, in cases presenting with chronic diarrhea, awareness of the CDDs should be increased and molecular analysis should be included in the diagnostic algorithm after carefully taken detailed history and routine examination. Thus, it will contribute to the reduction of morbidity and mortality rates by enabling CDD-specific treatment.
Conflict of Interest
No conflict of interest with any institution/organization.
References
- Kehar, Mohit (2016) Chronic diarrhea in children. Gastroenterology Hepatology Endoscopy: 40-43.
- (2013) World Health Organization: Diarrhoeal disease.
- Giannattasio, Antonietta; Guarino, Alfredo, Vecchio, et al. (2016) Management of children with prolonged diarrhea. F1000Researc 5.
- González Corona, Enrique Antonio (2017) Acute, prolonged and persistent diarrhea in children and its difference with chronic diarrhea. MediSan 21(09): 2047-2060.
- Roberto Berni Canani, Giuseppe Castaldo, Rosa Bacchetta, Martín G Martín, Olivier Goulet (2015) Congenital diarrhoeal disorders: advances in this evolving web of inherited enteropathies. Nature Reviews Gastroenterology & Hepatology 12(5): 293-302.
- Vincenza Pezzella, Giusi Grimaldi, Mariateresa Russo, Serena Mazza, Domenica Francesca Mariniello, et al. (2017) New insights and perspectives in congenital diarrheal disorders. Current Pediatrics Reports 5(3): 156-166.
- Ziqing Ye, Ying Huang, Cuifang Zheng, Yuhuan Wang, Junping Lu, et al. (2019) Clinical and genetic spectrum of children with congenital diarrhea and enteropathy in China. Genetics in Medicine 21(10): 2224-2230.
- Berni Canani R, Terrin G, Cardillo G, Tomaiuolo R, Castaldo G (2010) Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr 50: 360-366.
- Sharon Anderson, Soula Koniaris, Baozhong Xin, Susan Sklower Brooks (2017) Congenital glucose–galactose malabsorption: a case report. Journal of Pediatric Health Care 31(4): 506-510.
- Mehrin Sadiq, Omer Choudry, Arun K Kashyap, Danitza M Velazquez (2019) Congenital diarrhea in a newborn infant: A case report. World Journal of Clinical Pediatrics 8(3): 43.
- Philip A Kozan, Matthew D McGeough, Carla A Peña, James L Mueller, Kim E Barrett, et al. (2015) Mutation of EpCAM leads to intestinal barrier and ion transport dysfunction. Journal of molecular medicine 93(5): 535-545.
- Dilek Kahvecioğlu, Duran Yıldız, Atilla Kılıç, Banu İnce-Alkan, Ömer Erdeve, et al. (2014) A rare cause of congenital diarrhea in a Turkish newborn: tufting enteropathy. Turk J Pediatr 56(4): 440-443.
- Alexandre Fabre, Patrice Bourgeois, Marie-Edith Coste, Céline Roman, Vincent Barlogis, et al. (2017) Management of syndromic diarrhea/tricho-hepato-enteric syndrome: a review of the literature. Intractable & rare diseases research.
- Iddo Vardi, Ortal Bare, Michal Sperber, Michael Schvimer, Moran Nunberg, et al. (2018) Genetic and structural analysis of a SKIV2L mutation causing tricho-hepato-enteric syndrome. Digestive diseases and sciences 63(5): 1192-1199.
- Marta Germán-Díaz, Yolanda Rodriguez-Gil, Jaime Cruz-Rojo, Fabienne Charbit-Henrion, Nadine Cerf-Bensussan, et al. (2017) A new case of congenital malabsorptive diarrhea and diabetes secondary to mutant neurogenin-3. Pediatrics 140(2): e20162210.
- Nina L Gluchowski, Chandramohan Chitraju, Joseph A Picoraro, Niklas Mejhert, Shirly Pinto, et al. (2017) Identification and characterization of a novel DGAT1 missense mutation associated with congenital diarrhea. Journal of lipid research 58(6): 1230-1237.
- Thomas L Ratchford, Amelia J Kirby, Hailey Pinz, Dhiren R Patel (2018) Congenital diarrhea from DGAT1 mutation leading to electrolyte derangements, protein-losing enteropathy, and rickets. Journal of pediatric gastroenterology and nutrition 66(3): e82-e83.
- Hessa S Alsaif, Mohammad Al-Owain, Martin E Barrios-Llerena, Ghada Gosadi, Yousef Binamer, et al. (2019) Homozygous Loss-of-Function Mutations in AP1B1, Encoding Beta-1 Subunit of Adaptor-Related Protein Complex 1, Cause MEDNIK-like Syndrome. The American Journal of Human Genetics 105(5): 1016-1022.
- Incecik Faruk, Bisgin, Atil, Yilmaz, Mustafa (2018) MEDNIK syndrome with a frame shift causing mutation in AP1S1 gene and literature review of the clinical features. Metabolic brain disease 33(6): 2065-2068.
