 Case Report
Case ReportABSTRACT
Cutis Marmorata Telangiectatica Congenita is a very rare birth defect involving cutaneous blood vessels. Of unknown cause, uncertain pathophysiology, unclear epidemiology. Described as a localized, or generalized marbled skin appearance (cutis marmarota), in addition to the skin, it may involve any other body organs, with, or without a wide variety of associated congenital anomalies. Kato van Lohuizen described the first case in 1922. Since then, there have been less than 300 cases reported worldwide to date. We are adding one more case, and the first reported in Libya.
Keywords: Cutis Marmorata; Telangiectasia; Port-Wine Stains
Introduction
Van Lohuizen Syndrome, or Cutis Marmorata Telangiectatica Congenita. Is a very rare birth defect involving the cutaneous blood vessels, often reported as a benign, congenital disorder of unknown etiology, epidemiology, and pathophysiology. Described as persistent cutaneous telangiectasia, and phlebectasia, these appear as reticulated streaks of the skin capillaries and venules, resulting in a marbled-looking skin (cutis marmarota) [1-3]. First described by Kato van Lohuizen, a female Dutch pediatrician, in 1922 [4]. Since then, it has been referred to under several names, including, congenital generalized phlebectasia, [5,6] naevus vascularis reticularis, [7] as well as congenital livedo reticularis [8]. Characterized by the presence of erythematous network streaks, without venectasia, which is not responding to local heating. It may occur along with port-wine stain, [9] cutaneous ulceration, and atrophy within the affected area, as well as body asymmetry, and may affect any organ, including the eyes, skeleton, kidneys, and the brain [10-12].
Case Report
We are reporting the first case of CMTC in Libya, in 18 years old girl referred to our clinic for consultation. She had persistent skin disorders in her Rt. upper limb since birth (Figure 1). She is the third of three siblings, born as a full-term baby, by uncomplicated vaginal delivery, to a non-consanguineous marriage, and there was no family history of similar lesions. A high school student, leading a normal active life, until about a year ago, when she began to feel intermittently some discomfort and numbness of the right hand and forearm, with no history of trauma. Her upper extremity skin changes were detected at birth along with contralateral DDH, which was treated conservatively. Our clinical findings revealed a network of blue-purple lines giving the skin a marbled appearance along the right upper extremity from the deltoid insertion to the metacarpophalangeal joints level dorsally, sparing the Palmer skin, otherwise, her skin temperature and texture were normal, without atrophy or ulceration (Figure 2). The lesions were fading at Direct pressure, but not affected by gravity. There were multiple irregularly shaped port-wine stain patches on the right anckle laterally, left leg posteriorly, and the right thigh anteromedially. No upper or lower limbs atrophy, cercomfrential, or length discrepancy, and her gait was normal, there was no scoliosis, or facial asymmetry. There were no sensory or motor deficits detected clinically. Here skeletal survey, abdominal and pelvic ultrasound, echocardiography, and all laboratory investigations were within normal. NCS, reported mild right CTS, Based on this medical history and clinical presentation, the diagnosis of Van Lohuizen syndrome was confirmed (Figure 3).
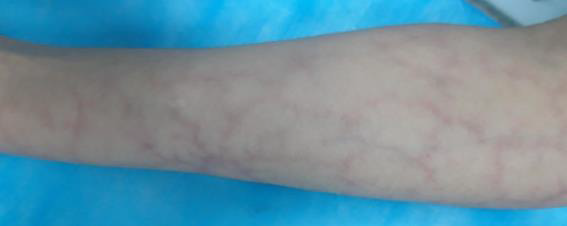
Figure 1: Cutis marmorata, Cutis marmorata, erythematous network of telangiectatic cutaneous capillaries.
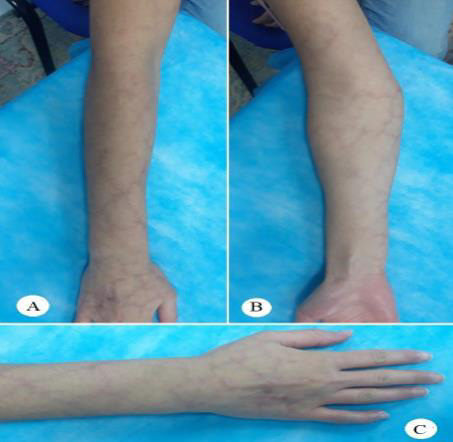
Figure 2: The lesion extends along the right upper limb, between the deltoid insertion proximally and the Metacarpophalangeal joints level distally, sparing the phalangeal skin dorsally and the palmar skin volarly.
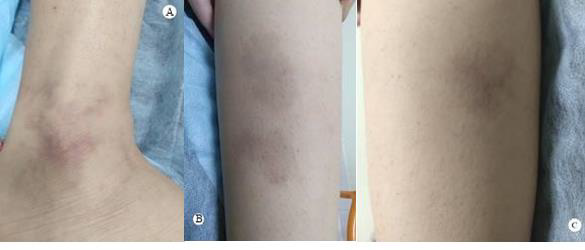
Figure 3: Multiple erregularly shaped port-wine stain patches on he Anckle A, Leg B and Thigh C.
Discussion
Van Lohuizen syndrome is a rare congenital disorder, with less than 300 cases reported worldwide to date [2,12,13]. However, since not all cases are diagnosed, it is difficult to determine incidence rates. Its origin and gender prevalence are still obscure and unclear, although, it is believed by some to be prevalent among females (64%) [14,15]. In some rare cases, CMTC may run in families [11]. However, Amitai et al. observed no familial cases of CMTC in their series, and most of the cases seem to be sporadic, with no racial predilection [16]. However, skin biopsies have revealed genetic mutations in GNA11, [17-20] and some researchers have also reported genetic mutations in ARL6IP6, suggesting its inheritance as a recessive trait [20-23]. Moreover, viral infections have been postulated, and the underlying connective tissue abnormality has been supposed. Its diagnosis is clinically based. It appears as cutaneous erythematous reticulum streaks, fading upon direct local pressure but unaffected by local heating or gravity, with no venectasia, findings that Kienast, [9] considered three of which as primary signs. While the presence of skin atrophy and ulceration, and the port-wine stains outside the areas affected by CMTC, have been considered a secondary diagnostic criterion (Table 1). For diagnosis, the presence of three major signs and two of the minor criteria is indicative [2].
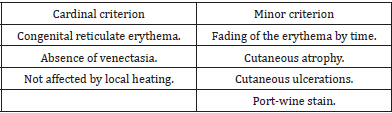
Table 1: The major and minor diagnostic criteria, Kienast classification [2].
In most cases (66.8%), the disorder is localized, and the leasions are unilateral in 65% of the cases, more frequently involving the limbs (69%), [16] where the upper limbs are affected in (25.9%), while the hands are involved in only (4.9%), but can be generalized in about (24.5%) of cases [24]. CMTC may involve any organ in the body, including the eyes, skeleton, brain, kidneys, and others [10]. The most frequently associated anomalies, in 18.8% of patients [16] include body asymmetry, neurological, ophthalmological, and cardiovascular defects, dysmorphic features, genitourinary, and endocrinological defects [2,16,24-29]. The CMTC should be distinguished from congenital livedo reticularis, which is caused by mosaic PIK3CA gene mutations, [30] where ulceration and phlebectasia do not occur, Sturge-Weber syndrome, which is caused by mosaic GNAQ gene mutations, [17] Neonatal lupus erythematosus, [31] and Klippel-Trenaunay syndrome.
Although, histopathology may confirm the diagnosis by showing an increased number and size of capillaries and veinules in the dermal layer, endothelial swelling, and sometimes aneurysms, it is not necessary, and nonspecific [16,32-35]. Imaging, and fluorescein angiography are indicated only for the evaluation of other congenital anomalies that may accompany the CMTC.
Up to the moment, there is no definitive treatment for skin symptoms in CMTC. However, sympathectomy, and laser therapy are postulated [14,32,34-38]. In our current case, we believe that treatment is unnecessary, apart from reassurance and may be a psychological support to ease the burden of the aesthetic appearance, for although the problem involves a wide area of about (9%) of the total body surface area (TBSA), [39] it is still localized, and it is without any other associated problem apart from the described port-wine patches. She is a tertiary school student, living a normal life with no functional or intellectual issues. However, if we consider her complaint of discomfort and numbness as part of the problem or some of its long-term sequels, [40] the clinical findings and investigation results do not suggest more than the need for follow-up. Although some authors, including Kienast, state that skin leasions typically become less and often completely disappear by adolescence, [3,4,16] this is not the case with our patient [12,41].
References
- Ozkur, Ezgi, Ilknur Kivanc Altunay, Tugba Falay Gur (2020) “What is your diagnosis?/Taniniz Nedir?." Turkish Archives of Dermatology and Venereology 54.3: 122-124.
- Kienast A K, P H Hoeger (2009) Cutis marmorata telangiectatica congenita: a prospective study of 27 cases and review of the literature with proposal of diagnostic criteria. Clinical and Experimental Dermatology: Clinical dermatology 34(3): 319-323.
- Proietti Ilaria, Nicoletta B, Veronica B, A Marchesiello, Sara Zuber, et al. (2020) Cutis Marmorata Telangiectatica Congenita: A Diagnostic Challenge. Giornale italiano di dermatologia e venereologia: organo ufficiale, Societa italiana di dermatologia e sifilografia 155(1): 108-110.
- Van Lohuizen CHJ (1992) "Uber eine seltene angeborene Hautanomalie (Cutis marmorata telangiectatica congenita)." Acta Derm Venereol 3: 202-211.
- Kantor I, D Yep (1966) "Congenital generalized phlebectasia." Archives of Dermatology 93(6) 515 N STATE ST, CHICAGO, IL 60610: AMER MEDICAL ASSOC.
- Lynch, Peter J, Alvin S Zelickson (1967) Congenital phlebectasia: a histopathologic study. Archives of dermatology 95(1): 98-101.
- Brain R T (1954) Naevus vascularis reticularis (two cases). Proc R Soc Med 47(3): 172-173.
- Champion R H (1965) "Livedo reticularis: A review." British Journal of Dermatology 77(4): 167-179.
- Rupprecht, Rainer, Max Hundeiker (1997) Cutis marmorata teleangiectatica congenita Wichtige Aspekte für die dermatologische Praxis. Der Hautarzt 48(1): 21-25.
- Powell S T, W P Su (1984) Cutis marmorata telangiectatica congenita: report of nine cases and review of the literature. Cutis 34(3): 305-312.
- Sybert, Virginia P (2017) Genetic skin disorders. Oxford University Press.
- Jia, Deng, Victor Samuel Rajadurai, Suresh Chandran (2018) Cutis marmorata telangiectatica congenita with skin ulceration: a rare benign skin vascular malformation. Case Reports bcr2018226763.
- Putkowski, Stefanie (2010) The National Organization for Rare Disorders (NORD) Providing Advocacy for People With Rare Disorders. NASN school nurse 25(1): 38-41.
- Elzouki, Abdelaziz Y (2011) Textbook of clinical pediatrics. Springer Science & Business Media.
- Pielop J A, M L Levy (2005) "Vascular lesions and congenital nevi in the newborn." UpToDate Online 13.
- Amitai, S Fichman, P Merlob, Y Morad, M Lapidoth, et al. (2000) Cutis marmorata telangiectatica congenita: clinical findings in 85 patients. Pediatr Dermatol 17(2): 100-104.
- Happle R (2015) Capillary malformations: a classification using specific names for specific skin disorders. Journal of the European Academy of Dermatology and Venereology 29(12): 2295-2305.
- Sassalos Thérèse M, T S Fields, Robert Levine, Hua Gao (2021) Retinal neovascularization from a patient with cutis marmorata telangiectatica congenita. Retinal Cases and Brief Reports 15(1): 77-80.
- Thomas Florian P, Velina Guergueltcheva, Francisco A A G, Ivailo Tournev, Chitharanjan V Rao, et al. (2016) Clinical, neurophysiological and morphological study of dominant intermediate Charcot-Marie-Tooth type C neuropathy. Journal of neurology 263(3): 467-476.
- Kumar Akash, Diane B Zastrow, Elijah J Kravets, Daniah Beleford, Maura R Z Ruzhnikov, et al. (2019) Extracutaneous manifestations in phacomatosis cesioflammea and cesiomarmorata: case series and literature review. American Journal of Medical Genetics Part A 179(6): 966-977.
- Dereure O (2016) Cutis marmorata telangiectatica congenita: mutations in a susceptibility gene involved in cerebrovascular accidents. Annales de dermatologie et de venereology 143(1): 96-97
- Abumansour Iman S, Hadia Hijazi, Anas Alazmi, Fatma Alzahrani, Fahad A Bashiri, et al. (2015) ARL6IP6, a susceptibility locus for ischemic stroke, is mutated in a patient with syndromic Cutis Marmorata Telangiectatica Congenita. Human genetics 134(8): 815-822.
- Happle Rudolf (1987) Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. Journal of the American Academy of Dermatology 16(4): 899-906.
- Bui, Teresa Nu Phuong Trinh, Ayse Corap, Anette Bygum (2019) "Cutis marmorata telangiectatica congenita: a literature review." Orphanet journal of rare diseases 14(1): 1-8.
- Devillers Arjan C A, Flora B de Waard-van der Spek, Arnold P Oranje (1999) Cutis marmorata telangiectatica congenita: clinical features in 35 cases. Archives of dermatology 135(1): 34-38.
- Gerritsen M J, P M Steijlen, H G Brunner, P Rieu P (2000) Cutis marmorata telangiectatica congenita: report of 18 cases. British Journal of Dermatology 142(2): 366-369.
- South David A, Alvin H Jacobs (1978) Cutis marmorata telangiectatica congenita (congenital generalized phlebectasia). The Journal of pediatrics 93(6): 944-949.
- Picascia, David D, Nancy B Esterly (1989) Cutis marmorata telangiectatica congenita: report of 22 cases. Journal of the American Academy of Dermatology 20(6): 1098-1104.
- Dar, Nasser Rashid, Tariq Ghafoor, and Zaheer Iqbal Awan. "Cutis marmorata telangiectatica congenita." Journal of the College of Physicians and Surgeons--pakistan: JCPSP 13(11): 653-655.
- Mirzaa Ghayda M, Robert L Conway, Karen W Gripp, Tally Lerman-Sagie, Dawn H Siegel, et al. (2012) Megalencephaly‐capillary malformation (MCAP) and megalencephaly‐polydactyly‐polymicrogyriahydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. American Journal of Medical Genetics Part A 158(2): 269-291.
- Del Boz Javier, María del Mar Serrano, Elisabeth Gómez, Ángel Vera (2009) Neonatal lupus erythematosus and cutis marmorata telangiectatica congenita‐like lesions. International journal of dermatology 48(11): 1206-1208.
- South David A, Alvin H Jacobs (1978) Cutis marmorata telangiectatica congenita (congenital generalized phlebectasia). The Journal of pediatrics 93(6): 944-949.
- Mazereeuw‐Hautier Juliette, Stephanie Carel‐Caneppele, JeanLouis Bonafé (2002) Cutis marmorata telangiectatica congenita: report of two persistent cases. Pediatric dermatology 19(6): 506-509.
- Hinek Aleksander, Shailly Jain, Glenn Taylor, David Nykanen, David Chitayat, et al. (2008) High copper levels and increased elastolysis in a patient with cutis marmorata teleangiectasia congenita. American Journal of Medical Genetics Part A 146(19): 2520-2527.
- Torrelo A, A Zambrano, R. Happle (2003) Cutis marmorata telangiectatica congenita and extensive mongolian spots: type 5 phacomatosis pigmentovascularis. British Journal of Dermatology 148(2): 342-345.
- Andreev V C, K Pramatarov (1979) Cutis marmorata telangiectatica congenita in two sisters. British Journal of Dermatology 101(3): 345-350.
- Deshpande Ajay J (2018) Cutis mormorata telangiectatica congenital successfully treated with intense pulsed light therapy: a case report. Journal of Cosmetic and Laser Therapy 20(3): 145-147.
- Sekido Mitsuru, Koji Adachi, Shinji Togashi, Kaoru Sasaki (2013) Laser therapy treatment of phacomatosis pigmentovascularis type II: two case reports.
- Livingston Edward H, Scott Lee (2000) Percentage of burned body surface area determination in obese and nonobese patients. Journal of surgical research 91(2): 106-110.
- Hu I-Jan, Ming-Ting Chen, Hao-Chih Tai, Po-Nien Tsao, Hung-Chieh Chou, et al. (2005) Cutis marmorata telangiectatica congenita with gangrenous ulceration and hypovolaemic shock. European journal of pediatrics 164(7): 411-413.
- https://rarediseases.org/rare-diseases/cutis-marmorata-telangiectatica-congenita/.
