 Mini Review
Mini ReviewAbstract
Until recently there were few options for the treatment of metastatic melanoma with chemotherapy agents (dacacarbazine and temozolamide), interleukin-2 therapy or combination biochemotherapy as the most common therapeutic approaches. Since 2007, 13 new FDA-approved therapies have been developed for treatment of melanoma. Here in we review the current and upcoming novel therapies in clinical development for the treatment of advanced and unresectable melanoma.
Keywords: HDAC Inhibitor; Melanoma; Epigenetic; Tucidinostat; HBI-8000
Abbreviations: ACS: American Cancer Society; AKT: Protein Kinase B; AML: Acute Myelogenous Leukemia; Anti-CTLA-4: Antibody Against Cytotoxic T-Lymphocyte- Associated Protein 4; Anti-PD-1: Antibody Against Programmed Cell Death-1 Protein 1; APL: Acute Promyelocytic Leukemia; ASCO: American Society of Clinical Oncology; ATLL: Adult T-Cell Leukemia/Lymphoma; Bcr Abl: Fusion of Bcr And Abl Genes; BEMPEG: Bempegaldesleukin; BIW: Twice A Week; BMS: Bristol Myers Squibb; Icpi: Immune Checkpoint Inhibitor; BRAF: V-Raf Murine Sarcoma Viral Oncogene Homolog B1; CTL: Cytotoxic T Lymphocyte; De Novo: ‘Of New’ Primary; DLBCL: Diffuse Large B-Cell Lymphoma; EMT: Epithelial Mesenchymal Transition; FDA: Food And Drug Administration; Flt-3: Fms-Like Tyrosine Kinase 3; HDAC: Histone Deacetylase; HER-2: Human Epidermal Growth Factor Receptor 2; HR: Hazard Ratio; HSP: Heat Shock Protein; IC50: Half-Maximal Inhibitory Concentration; IDMC: Independent Data Monitoring Committee; IDO: Indoleamine 2,3-Dioxygenase; IL-2: Interleukin-2; Iraes: Immune-Related Adverse Events; IT: Intratumoral; LAG-3: Lymphocyte-Activation Gene 3; MEK: Mitogen-Activated Extracellular Signal-Regulated Kinase; MHC: Major Histocompatibility Complex; MHLW: Ministry Of Health, Labor And Welfare; MTD: Maximum Tolerated Dose; NHL: Non-Hodgkin’s Lymphoma; NIH: National Institute Of Health; NKTR-214: Development Code For Bempegaldesleukin; NSCLC: Non-Small Cell Lung Cancer; Opdualag: Nivolumab/Relatlimab-Rmbw; ORR: Objective Response Rate; OS: Overall Survival; PD-1: Programmed Cell Death Protein 1; PD-L1: Programmed Cell Death-1 Ligand 1; PFS: Progression Free Survival; PRAME: Preferentially Expressed Antigen Of Melanoma; PTCL: Peripheral T-Cell Lymphoma; RCC: Renal Cell Carcinoma; R/R: Relapsed/Refractory; RP2D: Recommended Phase 2 Dose; SAHA: Suberoylanilide Hydroxamic Acid; SEER: Surveillance, Epidemiology, And Ends Results; Sirna: Small Interfering Ribonucleic Acid; SITC: Society For Immunotherapy of Cancer; SOC: Standard Of Care; TLR-9: Toll-Like Receptor-9; TME: Tumor Microenvironment; TVEC: Talimogene Laherparepvec; Treg: Regulatory T Cell; TSA: Trichostatin A; VEGF-R: Vascular Endothelial Growth Factor Receptor; VPA: Valproic Acid
Introduction
Metastatic melanoma is a serious form of invasive skin cancer, that affects adolescents and adults of all ages and is associated with a morbidity that has a substantial impact on day-to-day functioning with a highest risk of mortality in patients with unresectable and metastatic melanoma. The incidence of melanoma has doubled during the past three decades in the United States according to Center for Disease Control. The rate of new cases of melanoma of the skin is 22.8 per 100,000 men and women per year in the United States (US), and 2.3 percent of all adults will be diagnosed with melanoma of the skin in their lifetime. The percentage of melanoma cases observed by stage are 83% for localized, 9% regional, 4% distant, and 4% unknown. The 5-year relative survival rate from diagnosis for localized, early melanoma is over 99%, 68% for melanoma that has spread regionally, and 30% for melanoma that has spread to distant sites and 88% for unknown stage[1,2]. In the United States, estimated new cases for 2022 were 99,780 with 7,650 deaths [3].
Cancer Immunotherapy and Targeted Therapy in Melanoma
Currently, available therapies for metastatic melanoma include:
1) iCPI(s), nivolumab, pembrolizumab and anti-CTLA-4
ipilimumab;
2) Inhibitors to kinase pathways, including BRAF and mitogenactivated
protein kinase (MEK), vemurafenib, dabrafenib,
trametinib, cobimetinib, encorafenib, and binimetinib for
BRAF mutation-positive melanoma; and
3) Intratumoral (IT) vaccine therapy, TVEC (Imlygic™).
In the last decade immunotherapy with anti-PD-1 monoclonal antibodies (nivolumab and pembrolizumab) or anti-CTLA-4 antibody (ipilimumab) or targeted therapy for patients with BRAF mutated tumor has demonstrated prolonged survival compared to earlier approaches. The antibodies directed against the PD-1 receptor (OPDIVO®, Keytruda®) or its ligand, PD-L1 (Tecentriq®), target this immune checkpoint interaction and at least in a subset of melanoma of patients, promote antitumor immunity through inhibition of the PD-1/PD-L1 checkpoint signaling axis. These new treatment options have led to a marked improvement in survival of patients with metastatic melanoma, however, even with the current optimal therapy approximately 50% of patients diagnosed with metastatic melanoma will succumb to death within 5 years [4]. Until recently, only anti-CTLA-4 has shown an incremental benefit when added to anti-PD-1 antibody but has also resulted in a substantial increase in toxicity. The combination of nivolumab plus ipilimumab showed an absolute increase in survival of 6% (52% to 58%) at 3 years compared to nivolumab alone but with an increase in treatment-related Grade 3 or 4 adverse events from 21% to 59% [4]. Several new agents in combination with anti- PD-1 immunotherapy are being examined in clinical studies with the objective of increasing response rate and improving survival with an acceptable safety and tolerability. These include, but not limited to, indoleamine 2,3-dioxygenase (IDO) inhibitors, Toll-like receptor-9 (TLR-9) agonists, pegylated interleukin (IL-2) and antilymphocyte activation gene (LAG)-3 antibody [5,6].
The ongoing clinical trials testing different combination therapy with immune checkpoint inhibitor in advanced melanoma showed mixed results. The Phase 3 PIVOT IO-001 study evaluating the doublet therapy of Nektar Therapeutic’s bempegaldesleukin (BEMPEG/NKTR-214) in combination with Bristol-Myers Squib (BMS) Opdivo (nivolumab) compared to Opdivo monotherapy as a first-line treatment for previously untreated unresectable or metastatic melanoma - PIVOT-12 trial (NCT04410445) did not meet its primary endpoints [7]. Following a review of the study for efficacy and safety by an independent Data Monitoring Committee (IDMC), the study did not meet the primary endpoints of progression free survival (PFS) and objective response rate (ORR) and the third primary endpoint of overall survival (OS) also did not meet statistical significance at the first interim analysis. RELATIVITY-047, a global Phase III trial testing the novel immunotherapeutic combination of nivolumab + relatlimab which targets PD-1 and LAG-3 pathways on the other hand met its primary endpoint of PFS in extending progression free survival as a first-line treatment of advanced or unresectable melanoma [8]. After a median follow-up of 13.2 months, the study met its primary endpoint by significantly improving median progression-free survival vs nivolumab alone. By blinded independent review, median progression-free survival was 10.2 months vs 4.6 months (hazard ratio [HR] = 0.75, P = .0055). Opdualag, a fixed-dose combination of nivolumab 480 mg + relatlimab 160 mg administered intravenously every 4 weeks was approved by FDA on March 18, 2022 although the secondary endpoint of median overall survival was not reached with this combination which was 34.1 months with nivolumab alone (HR = 0.80, P = .0593). Thus, Opdualag did not show any overall survival benefit.
Despite new treatment options, there remains a substantial unmet need for treatments that extend survival and provide a better quality of life for patients with advanced and unresectable melanoma. Anti-PD-1 and anti-cytotoxic T-lymphocyte-associated protein-4 (anti-CTLA-4) therapies are constrained owing to primary (de novo) and acquired resistance. This resistance is attributed in part to epigenetic alterations in cells of the tumor microenvironment (TME) that play a major role in creating an immunosuppressive environment and a lack of effectiveness of immune checkpoint inhibitors (iCPIs) therapies [9]. Additionally, combination therapy with nivolumab and ipilimumab results in high grade toxicity rates of 55% to 59% including immunerelated adverse events (irAEs) that result in dose reductions and discontinuation of treatment [10]. Approved standard of care (SOC) therapies have limited ability to treat metastatic melanoma due to significant heterogeneity in response and toxicity, and, as such, these SOC treatments do not adequately address the medical needs of all patients with metastatic melanoma.
Histone Deacetylases as a Therapeutic Target in Cancer
Histone acetylation and deacetylation play important roles in the modulation of chromatin topology and the regulation of gene transcription. There are three classes of histone deacetylase (HDAC) enzymes (I, IIa/IIb, and IV) that utilize a zinc catalyzed mechanism for deacetylation of histones and non histone proteins [11-17]. The HDAC inhibitors are a class of epigenetic modulators that act as cytostatic agents to inhibit the proliferation of certain type of cancer cells in culture and in vivo by inducing cell cycle arrest and apoptosis [18]. HDAC activity is increased in several cancers and may promote the malignant state through a variety of mechanisms. Expression or activity of Class I HDACs (HDAC1, HDAC2, HDAC3, and HDAC8) is elevated in acute myelogenous leukemia (AML); acute promyelocytic leukemia (APL); non-Hodgkin’s lymphoma (NHL); and prostate, gastric, colorectal, breast, and cervical cancers [11,19-22]. Small interfering ribonucleic acid (siRNA) mediated inhibition of HDAC1 or HDAC3 resulted in anti proliferative effects, and HDAC2 inhibition using siRNA sensitizes tumor cells to apoptosis [16,23,24].
Class IIb HDACs (HDAC6 and HDAC10) have been shown to preferentially target non histone proteins, such as alpha tubulin and heat shock protein (HSP) 90. HSP90 has been shown to participate in malignant transformation by stabilizing oncoproteins such as Bcr Abl, mutant flt-3, AKT, c-Raf, estrogen receptors, HER-2, and vascular endothelial growth factor receptor VEGF-R. Alpha tubulin suppresses apoptosis in tumor cells by facilitating lysosomal clearance of misfolded proteins. Deacetylation of HSP90 and alpha tubulin by HDAC6 or HDAC10 activates these proteins, contributing to the malignant phenotype. Conversely, inhibitions of HDAC6 or HDAC10 have been shown to inhibit tumor growth. The synergy observed with the HSP90 inhibitor 17-AAG and the antitumor proteasome inhibitor bortezomib further support these proposed mechanisms [25-29]. Given the strong association of HDAC activity with cancer, inhibition of HDACs has represented a promising therapeutic strategy for cancer treatment.
Rationale for Use of Selective HDAC Inhibitors for the Treatment of Cancer
Cancer may be caused by genetic defects such as gene mutations and abnormal gene expression. Epigenetic dysregulation, is directly contributing in the growth of cancer cells and also affect immunocyte function [30]. Some of these cancer cell intrinsic defects are reversible and the promise of epigenetic regulators like HDAC inhibitors is that they can concurrently target multiple aberrant or compensatory signaling pathways found in cancer cells by restoring the genome, and by modulation of transcriptome, to a more normal like state [31]. Experiments performed with human lymphoblastoid cell lines showed genes in several functional clusters such as lysine acetyltransferase (KAT) were down regulated as well as genes required for growth and maintenance of the lymphoid phenotype. Whereas up-regulated gene clusters were enriched in regulators of transcription, development and phenotypic change were regulated by Class I HDAC Inhibitors such as vorinostat [32]. This includes several genes important for cell cycle (CCNA2/Cyclin A2, CCNB2/Cyclin B2, CCNE2/Cyclin E2, CDKN1A/p21/WAF1, CHEK1/checkpoint kinase 1), apoptosis (DR6/TNFRSF21/death receptor 6) and the response to antitumor therapies (ABCB10, ABCC2/MRP2, RAD23B, UBCH10/ubiquitin conjugating enzyme E2C) [33,34]. Tucidinostat also regulated genes promoting epithelial differentiation (CDH1, KRT8) and reducing the epithelial mesenchymal transition (EMT), an important process in tumor invasion and metastasis (CDH2/N cadherin) [35]. In addition to HDAC1, 2, and 3, specific inhibition of HDAC6 or HDAC10 has been shown to reduce expression of VEGF R1 and VEGF R2 in tumor cells, presumably via enhancement of HSP90 acetylation suggesting an additional mechanism for anti-tumor activity of HDACs [27].
Role of HDAC Inhibitors in Tumor Immunology
The unique aspects of HDAC inhibition that contribute to anticancer activity have been described, e.g., reprogramming distorted transcription patterns and signal pathways in tumor cells, and modulation of the tumor microenvironment (TME) via induction of the activity of the immune system [12,36]. For instance, HDAC inhibitors of the benzamide class induce major histocompatibility complex (MHC) Class I like antigens (e.g., MHC 1 polypeptide-related sequence A [MICA], MICB) and the ligand of the natural killer (NK) group 2D protein (NKG2D), which can stimulate innate antitumor immunity [37]. Similar effects were observed with tucidinostat on in vitro cell lines and in clinical samples [33]. Tucidinostat enhances the cytotoxic effects of human peripheral mononuclear cells ex vivo on K562 target cells via upregulation of proteins involved in NK cell functions (e.g., NKG2D and granzymes). This NK activity was also observed in peripheral white blood cells derived from 2 lymphoma patients who responded to chidamide administration [33].
It has been reported that chidamide induces expression of the leukemia specific antigen preferentially expressed antigen in melanoma (PRAME) in both cell lines and leukemia blasts in bone marrow samples from patients, resulting in increased PRAME specific and cytotoxic T lymphocyte (CTL) mediated in vitro cytotoxicity against leukemia [38]. Most importantly, the pharmacological concentrations required to activate either NK mediated or antigen specific CTL activities by tucidinostat in vitro, ex vivo and in patients were between 100 and 500 nM, a concentration where cytotoxicity against normal cells was not evident. Inhibition of different subtypes of HDACs could have distinct effects on regulatory T cell (TREG) activity. For example, inhibition of Class I, Class II or Class IV subtypes 6, 9, and 11 may exaggerate TREG expansion while inhibition of Class I subtypes 1, 2, and 3 may repress the TREG expansion, which in general favors the antitumor immune response by Class I selective inhibitors [39-42].
In summary, emerging data suggest that there are two distinct mechanisms of action associated with tucidinostat, a direct antitumor mechanism and immunomodulatory mechanism both of which occur as a consequence of epigenetic modulation [33,34]. The direct antitumor mechanism is mediated by preferential induction of growth arrest and apoptosis in blood and lymphoid derived tumor cells. The immunomodulatory mechanism is mediated by activation of NK mediated and CD8+ CTL mediated antigen specific cellular antitumor activity leading to partial reversal of EMT and drug resistance of tumor cells. Tucidinostat (also known as chidamide, HBI-8000, CS055, Epidaza®, Hiyasta®) is an epigenetic regulator that can concurrently target multiple aberrant, silenced, overexpressed, or other compensatory signaling pathways found in cancer cells [43,44]. Tucidinostat displays a gene regulation pattern that is consistent with that hypothesis [45].
In contrast, pan HDAC inhibitors (e.g., valproic acid [VPA], trichostatin A [TSA], and suberoylanilide hydroxamic acid [SAHA]) have been reported to inhibit both NK and CTL activity, and are less desirable in cancer immunotherapy where enhancement of antitumor immune response is essential for efficacy [46]. Therefore, selective HDAC inhibitors such as tucidinostat are considered more suitable in cancer immunotherapy [47-49]. Immunomodulatory effects of tucidinostat such as activation of NK and CD8 T cell mediated antitumor activity makes it an attractive candidate HDAC inhibitor for combination therapy with immune checkpoint inhibitors (iCPI), such as anti PD-1 antibodies [45].
Rationale for Using Tucidinostat and CPI
Inhibition of different subtypes of HDACs could have distinct effects on TREG activity [39-42]. Furthermore, a recent publication demonstrated the newly discovered role of HDAC2 in the nuclear translocation of PD-L1 that regulates the immune response gene expression [50]. This investigation is consistent with the observation that inhibition of HDAC2 dependent acetylation of intracellular transport system could enhance the therapeutic effect of immune checkpoint inhibitors commonly used in the clinic. Recent reports suggest that HDAC inhibition has a significant effect on the expression of immune checkpoint co-inhibitory and co-stimulatory molecules. Additionally, HDAC inhibition may affect immunogenicity, antigen-presenting cell and T cell priming, regulatory T cells, myeloid-derived suppressor cells, and effector cell functions [51-54]. For example, HDAC inhibition has been shown to upregulate PD-1 ligand in melanoma and thereby augmenting immunotherapy with PD-1 blockade [55]. Class I HDAC inhibitors upregulate the expression of PD-L1 and to a lesser extent PD L2 in human and murine melanoma cell lines and in tumor tissue isolated from cancer patients.
The upregulation of PD-1 ligands was durable and lasted beyond 96 hours. These results suggest that combination of Class I HDAC inhibitors, such as tucidinostat, may have additive or synergistic activity in combination with PD-1 blockade antibodies. The direct evidence supporting the synergy of tucidinostat with immune checkpoint inhibitors was observed in preclinical studies. Combination of tucidinostat and anti-PD-1 mAb significantly inhibited growth of MC38 tumors compared to either single agent therapy. Gene expression analysis of the treated MC38 tumors revealed significant changes in the expression of immune checkpoints, with enhanced dendritic cell and antigen-presenting cell functions, and modulation of MHC class I and II molecules. These findings suggest that tucidinostat mediates epigenetic modifications in the tumor microenvironment, leading to improved efficacy of immune checkpoint inhibitors, and provide strong rationale for combination therapies (tucidinostat plus CPI) in the clinical setting [45].
Rationale for the Use of HDAC Inhibitors in Melanoma
Investigating antimelanoma potential of HDAC inhibitors have indicated that HDAC inhibitors suppress growth of melanoma cells by affecting not only growth/survival, but also by increasing their immunogenicity [55-57]. These data provide a scientific rationale for use of HDAC inhibitors in combination with CPIs in melanoma patients [57]. It was shown that treatment with HDAC inhibitor such as panobinostat (LBH589) results in increased expression of pro inflammatory surface markers on melanoma cells and increased surface expression of MHC, CD40, and CD80, thus increasing the immunogenicity of melanoma cells and promoting enhanced T cell activation [55,57,58]. These results indicate that HDAC inhibitors are a potentially useful class of compounds for elucidating cell signaling pathways and for developing new approaches to the treatment of melanoma [56]. Combination of HDAC inhibitor MS-275 and interleukin 2 (IL 2) increased antitumor effect in a melanoma model via activated cytotoxic T cells [46]. Furthermore, recent clinical study data has shown that blockade of the PD-1/PDL1 interaction is effective in the treatment of melanoma, renal cell carcinoma (RCC), and non small cell lung cancer (NSCLC) [59,60].
Importantly, responses to PD-1 blocking antibodies were preferentially seen in patients with tumors expressing PD-L1, and HDAC inhibitors targeting Class I HDACs, but not Class II, augmented expression of PD-L1 in melanoma cells [55,57]. In addition, increased expression of PD-L1 is dependent on inhibition of multiple Class I HDACs. Class I HDAC inhibition upregulates PD-1 ligands in melanoma and increases the efficacy of PD-1 blockade. Combination of an HDAC inhibitor and anti-PD-L1 monoclonal antibody in a preclinical model of melanoma showed a survival advantage over those receiving either single agent therapy [61]. It was also shown that HDAC inhibitors sensitize apoptosis resistant melanomas to human CTLs through regulation of TRAIL/DR5 pathway [62,63]. HDACs acting as mediators of resistance to apoptosis in melanoma and as targets for combination therapy with selective rapid acceleration of fibrosarcoma gene B (BRAF) inhibitors; co targeting of HDACs and oncogenic BRAF synergistically killed human melanoma cells [64,65].
Tucidinostat (HBI-8000): A Class I HDAC Inhibitor
Tucidinostat is a member of the benzamide class of HDAC inhibitors designed to block the catalytic pocket of Class I HDACs. In biochemical assays, tucidonostat inhibits several cancer associated Class I HDACs and one Class IIb HDAC in the nanomolar range and stimulates accumulation of acetylated histones H3 and H4 in tumor cells [33]. In vitro, Tucidinostat inhibits the growth of a variety of tumor cell lines, with half maximal inhibitory concentrations (IC50) in the single digit micromolar range and was non toxic to non transformed cells (IC50 ≥ 100 micromolar) [33]. In vivo, tucidinostat has demonstrated dose dependent antitumor activity against a panel of mouse and human xenograft models, producing 25% to 84% inhibition of tumor growth at doses that did not cause weight loss or any overt signs of toxicity [33].
HUYABIO International, LLC (HUYABIO™) has licensed worldwide rights outside China to the compound and is currently developing HBI-8000 in combinations for the treatment of solid tumors (such as melanoma, breast, kidney, and lung cancers) and in diffuse large B-cell lymphoma (DLBCL). HUYABIO is developing HBI-8000 as a monotherapy for treatment of hematological malignancies and has received approval in Japan. The Ministry of Health, Labor and Welfare (MHLW) in Japan approved tucidinostat 10 mg tablet for treatment of R/R adult T-cell leukemia/lymphoma (ATLL) on 23 Jun 2021 and additional indication for R/R peripheral T cell lymphoma (PTCL) on 25 Nov 2021. To date, multiple clinical trials that have been initiated and/or completed with tucidinostat (HBI-8000) in patients with various types of cancer by HUYABIO, as of data cut-off date of 22 Dec 2021, is provided in Table 1.
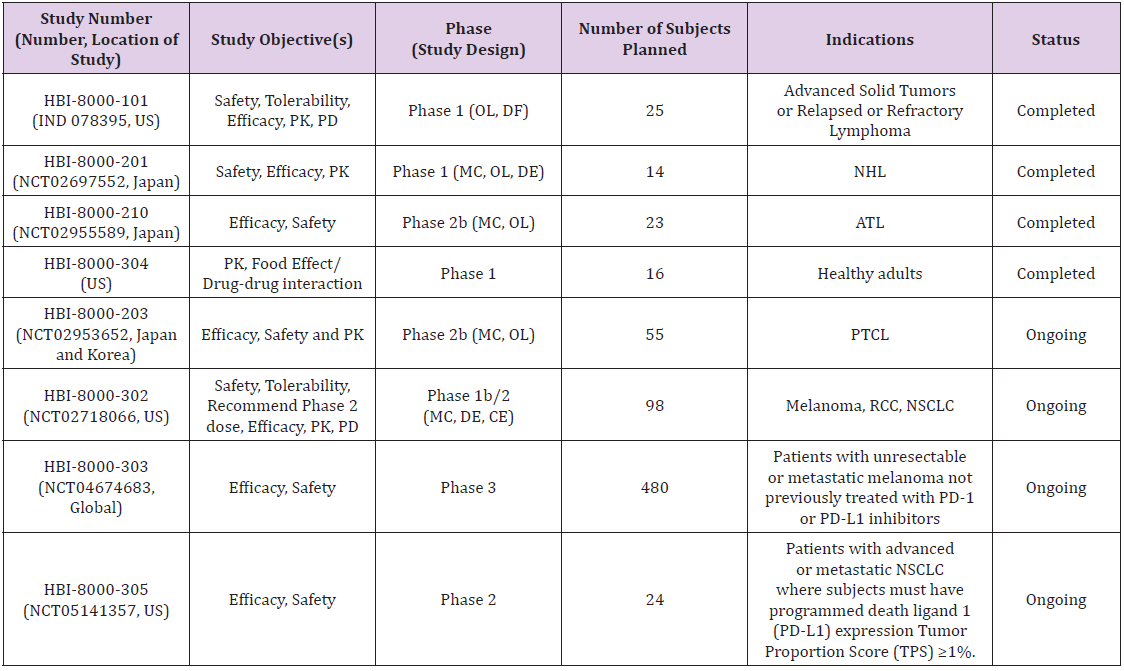
Table 1: Clinical Trials Conducted by HUYABIO International, LLC with HBI-80000 (tucidinostat).
Note: ATL = adult Tcell lymphoma; CE = cohort expansion; DE = dose-escalation; DF = dose-finding; IV = intravenous; MC = multicenter; NHL = nonHodgkin’s lymphoma; NSCLC = nonsmall cell lung cancer; OL = open label; PD = pharmacodynamics; PD1 = programmed cell death protein 1; PD-L1 = programmed deathligand 1; PK = pharmacokinetic; PTCL = peripheral Tcell lymphoma; RCC = renal cell carcinoma; TPS = tumor proportion score; US = United States.
Ongoing Clinical Trials with Tucidinostat
Phase 1b/2 Study (HBI-8000-302) in Solid Tumors (Melanoma, Non-Small Cell Lung Cancer and Renal Cell Carcinoma): HBI-8000-302 is an ongoing US Phase 1b/2 clinical trial open-label testing tucidinostat in combination with nivolumab conducted in patients with advanced solid tumors including melanoma (n=56), renal cell carcinoma (RCC) (n=11), and NSCLC (n=29). The primary objective of this Phase 1b/2 study was to evaluate the safety and tolerability of tucidinostat when combined with a standard dose and regiment of nivolumab, determine a maximum tolerated dose (MTD) and/or recommended Phase 2 dose (RP2D) and to evaluate frequency and severity of toxicities of this combination treatment. Three dose levels of tucidinostat (with nivolumab administered at 240 mg by IV infusion every 2 weeks), 20 mg BIW, 30 mg BIW, and 40 mg BIW were planned for the Phase 1b part of the study. The decision on dose escalation was based on the conventional 3+3 design and observed incidences of DLT. The Phase 1b dose finding part of the study has completed with determination of the recommended Phase 2 dose as 30mg BIW. The Phase 2 dose expansion study to explore the preliminary efficacy of tucidinostat at MTD and/or RP2D in combination with nivolumab is ongoing [66]. Preliminary data on efficacy and safety of this combination was presented at SITC 2020 Annual Meeting [67]. which showed that the combination of HBI-8000 30mg BIW and nivolumab was well tolerated and demonstrated encouraging efficacy and safety in patients with anti-PD1-naïve advanced melanoma. Further investigation of this promising combination regimen in the Phase 3 setting is currently being activated comparing Nivolumab alone to Nivolumab plus HBI-8000.
Phase 3 Double Blind HBI-8000-303 Clinical Trial in Patients with Unresectable or Metastatic Melanoma Not Previously Treated with PD-1 or PD-L1 Inhibitors: Based on significant anti-tumor activity and well tolerated safety profile generated in the Phase 2 clinical trial (HBI-8000-302) data in checkpoint naïve unresectable or metastatic melanoma, HUYABIO International, LLC initiated HBI-8000-303 global Phase 3 pivotal clinical trial. This is an ongoing multicenter, randomized, doubleblind Phase 3 Study of tucidinostat combined with nivolumab versus placebo with nivolumab in patients with unresectable or metastatic melanoma not previously treated with PD-1 or PDL1 inhibitors. This study includes a special open label cohort of approximately 30 patients who are either adult (or adolescents, 12 to 17 years of age in countries where approved) with new, progressive brain metastasis.
Discussion
The onset of cancer may arise via genetic alteration (mutations or deletion) and or via epigenetic modulation or silencing which can occur with deregulation of the epigenetic machinery by methylation of CpG sequence in promoter/enhancer regions that governs gene expression or alterations in histone posttranslational modifications or protein binding to chromatin [32]. This has led to the realization that epigenetic modulation of cancer-related genes by HDAC inhibitors may provide therapeutic treatment for a variety of cancers [68]. HDAC inhibitors as single agent did not show significant anti-tumor activities especially in solid tumors [69]. HDAC inhibitors in combination with chemotherapy and immune- therapies have demonstrated utility in reversing treatment resistance and sensitize cancer cells to further therapeutic interventions [70]. Preclinical and early clinical trials with tucidinostat have demonstrated significant anti-tumor activity in combination with anti-PD-1 with well tolerated safety profile generated in the Phase 2 clinical trial (HBI-8000-302) data in checkpoint naïve unresectable or metastatic melanoma, HUYABIO International, LLC initiated HBI-8000-303 global Phase 3 pivotal clinical trial to bring this novel combination therapy to meet unmet medical needs in advanced and unresectable melanoma patients.
Conclusion
Recent advances in the treatment of advanced and unresectable melanoma demonstrate that despite the dramatic improvements in the outcomes in the wake of US Food and Drug Administration (FDA) approval of new therapies for metastatic disease, about half of the patients do not respond to frontline therapy or relapse within months of initiating treatment [52,71]. In addition, treatment toxicities, particularly immune-related toxicities, often limit the applicability of these treatments to all patients with melanoma. Currently, there is no cure for advanced unresectable or metastatic melanoma. Therefore, an unmet medical need for new therapies remains to be developed. HBI-8000 in combination with nivolumab demonstrates a promising response and acceptable toxicity profile for the treatment of patients with unresectable or metastatic melanoma not previously treated with PD-1 or PD-L1 inhibitors. We eagerly await the results of HBI-8000-303 clinical trial [72] for potentially more efficacious combination treatment for advanced and unresectable melanoma patients along with better quality of life.
Acknowledgment
Medical writing assistance was provided by Christina Fleming, PhD. This support was funded by HUYABIO International, LLC.
References
- Melanoma Research Alliance, accessed March 30, 2022.
- NIH SEER 2021. National Cancer Institute-Surveillance, Epidemiology, and End Results Program.
- (2022) ACS. Cancer Facts & Figures, accessed March 30, 2022.
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, et al. (2017) Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 377(14): 1345-1356.
- Ascierto PA, Flaherty K, Goff S (2018) Emerging strategies in systemic therapy for the treatment of melanoma. American Society of Clinical Oncology Educational Book 38: 751-758.
- Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, et al. (2022) Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med 386: 24-34.
- PIVOT-12 Phase 3 trial of combination for advanced melanoma misses primary endpoints.
- Helwick C (2022) Immunotherapy duo delays disease progression in previously untreated patients with melanoma: RELATIVITY-047. ASCO Post March 16, 2022.
- Lodewijk I, Nunes SP, Henrique R, Jeronimo C, Duenas M, et al. (2021) Tackling tumor microenvironment through epigenetic tools to improve cancer immunotherapy. Clin Epigenetics 13(1): 63.
- Moreira A, Heinzerling L, Bhardwaj N, Friedlander P (2021) Current melanoma treatments: where do we stand? Cancers (Basel) 13(2): 221.
- Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5(9): 769-784.
- Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6(1): 38-51.
- Glaser KB (2007) HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol 74(5): 659-671.
- Rasheed W, Johnstone RW, Prince HM (2007) Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs 16(5): 659-678.
- Rasheed W, Bishton M, Johnstone RW, Prince HM (2008) Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev Anticancer Ther 8(3): 413-432.
- Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10(1): 32-42.
- Witt O, Deubzer HE, Milde T, Oehme I (2009) HDAC family: What are the cancer relevant targets? Cancer Lett 277(1): 8-21.
- Yang L, Liang Q, Shen K, Ma L, An N, et al. (2015) A novel class I histone deacetylase inhibitor, I-7ab, induces apoptosis and arrests cell cycle progression in human colorectal cancer cells. Biomed Pharmacother 71: 70-78.
- Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, et al. (2007)Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep 18(4): 769-774.
- Fritzsche FR, Weichert W, Roske A, Gekeler V, Beckers T, et al. (2008) Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer 8: 381.
- Weichert W, Denkert C, Noske A, Darb-Esfahani S, Dietel M, et al. (2008a) Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia 10(9): 1021-1027.
- Weichert W, Roske A, Gekeler V, Beckers T, Stephan C, et al. (2008b) Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer 98(3): 604-610.
- Inoue S, Mai A, Dyer MJ, Cohen GM (2006) Inhibition of histone deacetylase class I but not class II is critical for the sensitization of leukemic cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res 66(13): 6785-6792.
- Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, et al. (2008) Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell 30(1): 61-72.
- Glaser KB, Li J, Pease LJ, Staver MJ, Marcotte PA, et al. (2004) Differential protein acetylation induced by novel histone deacetylase inhibitors. Biochem Biophys Res Commun 325(3): 683-690.
- Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, et al. (2005) Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 280(29): 26729-26734.
- Park JH, Kim SH, Choi MC, Lee J, Oh DY, et al. (2008) Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem Biophys Res Commun 368(2): 318-322.
- Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu, et al. (2008) Role of the aggresome pathway in cancer: targeting histone deacetylase 6–dependent protein degradation. Cancer Research 68(8): 2557-2560.
- Schemies J, Sippl W, Jung M (2009) Histone deacetylase inhibitors that target tubulin. Cancer Lett 280(2): 222-232.
- Baylin SB, Jones PA (2016) Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 8: a019505.
- Li Y, Seto E (2016) HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med 6: a026831.
- Halsall JA, Turan N, Wiersma M, Turner BM (2015) Cells adapt to the epigenomic disruption caused by histone deacetylase inhibitors through a coordinated, chromatin-mediated transcriptional response. Epigenetics & Chromatin 8: 29-45.
- Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, et al. (2012) Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol 69(4): 901-909.
- Pan DS, Yang QJ, Fu X, Shan S, Zhu J-Z, et al. (2014) Discovery of an orally active subtype-selective HDAC inhibitor, chidamide, as an epigenetic modulator for cancer treatment. MedChemComm 5(12): 1789-1796.
- Meiying JI, Lee EJ, Kim KB, Kim Y, Sung R, et al. (2015) HDAC inhibitors induce epithelial-mesenchymal transition in colon carcinoma cells. Oncol Report 33: 2299-2308.
- Pattabiraman DR, Weinberg RA (2014) Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov 13(7): 497-512.
- Schmudde M, Braun A, Pende D, Sonnemann J, Klier U, et al. (2008) Histone deacetylase inhibitors sensitize tumour cells for cytotoxic effects of natural killer cells. Cancer Lett 272(1): 110-121.
- Yao Y, Zhou J, Wang L, Gao X, Ning Q, et al. (2013) Increased PRAME-specific CTL killing of acute myeloid leukemia cells by either a novel histone deacetylase inhibitor chidamide alone or combined treatment with decitabine. PLoS One 8(8): e70522.
- Wang L, Tao R, Hancock WW (2009) Using histone deacetylase inhibitors to enhance Foxp3(+) regulatory T-cell function and induce allograft tolerance. Immunol Cell Biol 87(3): 195-202.
- Beier UH, Wang L, Han R, Akimova T, Liu, et al. (2012) Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal 5(229): ra45.
- Pili R, Salumbides B, Zhao M, Altiok S, Qian D, et al. (2012) Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer 106(1): 77-84.
- Shen L, Pili R (2012) Class I histone deacetylase inhibition is a novel mechanism to target regulatory T cells in immunotherapy. Oncoimmunology 1(6): 948-950.
- Zhao R, Choi BY, Lee MH, Bode AM, Dong Z (2016) Implications of genetic and epigenetic alterations of CDKN2A (p16INK4a) in cancer. E Bio Medicine 8: 30-39.
- Zhang L, Chen Y, Jiang Q, Song W, Zhang L, et al. (2019) Therapeutic potential of selective histone deacetylase 3 inhibition. Eu J Med Chem 162: 534-542.
- Bissonnette RP, Cesario MC, Goodenow B, Shojaei F, Gillings M (2021) The epigenetic immunomodulator, HBI-8000, enhances the response and reverses resistance to checkpoint inhibitors. BMC Cancer 21: 969-986.
- Kato Y, Yoshino I, Egusa C, Maeda T, Pili R, et al. (2014)Combination of HDAC inhibitor MS-275 and IL-2 increased anti-tumor effect in a melanoma model via activated cytotoxic T cells. J Dermatol Sci 75(2): 140-147.
- Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl J (2007) Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett 581(7): 1317-1322.
- Rossi LE, Avila DE, Spallanzani RG, Ziblat A, Fuertes, et al. (2012) Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J Leukoc Biol 91(2): 321-331.
- Jones RB, O'Connor R, Mueller S, Foley M, Szeto GL, et al. (2014) Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog 10(8): e1004287.
- Gao Y, Nihira NT, Bu X, Chu C, Zhang J, et al. (2020) Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol 22(9): 1064-1075.
- Oronsky B, Oronsky N, Knox S, Fanger G, Scicinski J (2014) Episensitization: therapeutic tumor resensitization by epigenetic agents: a review and reassessment. Anticancer Agents Med Chem 14(8): 1121-1127.
- Briere D, Sudhakar N, Woods DM, Hallin J, Engstrom LD, et al. (2018) The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol Immunother 67(3): 381-392.
- Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, et al. (2018) Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res 6(12): 1561-1577.
- Bretz AC, Parnitzke U, Kronthaler K, Dreker T, Bartz R, et al. (2019) Domatinostat favors the immunotherapy response by modulating the tumor immune microenvironment (TIME). J Immunother Cancer 7(1): 294.
- Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor E, et al. (2015)HDAC Inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res 3(12): 1375-1385.
- Boyle GM, Martyn AC, Parsons PG (2005) Histone deacetylase inhibitors and malignant melanoma. Pigment Cell Res 18(3): 160-166.
- Woods DM, Woan K, Cheng F, Wang H, Perez-Villarroel P, et al. (2013) The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res 23(5): 341-348.
- Dokmanovic M, Clarke C, Marks PA (2007) Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 5(10): 981-989.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, et al. (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366(26): 2443-2454.
- Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, et al. (2014) Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 32(10): 1020-1030.
- Woods DM, Sodre AL, Sahakian E, Powers J, Lienlaf-Moreno M, et al. (2014) Abstract 4090: Inhibition of class I histone deacetylases promotes robust and durable enhancement of PDL1 expression in melanoma: Rationale for combination therapy. Cancer Research 74(19 Supplement): 4090-4090.
- Jazirehi AR, Kurdistani SK, Economou JS (2014) Histone deacetylase inhibitor sensitizes apoptosis-resistant melanomas to cytotoxic human T lymphocytes through regulation of TRAIL/DR5 pathway. J Immunol 192(8): 3981-3989.
- Balliu M, Guandalini L, Romanelli MN, D'Amico M, Paoletti F (2015) HDAC-inhibitor (S)-8 disrupts HDAC6-PP1 complex prompting A375 melanoma cell growth arrest and apoptosis. J Cell Mol Med 19(1): 143-154.
- Lai F, Jin L, Gallagher S, Mijatov B, Zhang X, et al. (2012) Histone deacetylases (HDACs) as mediators of resistance to apoptosis in melanoma and as targets for combination therapy with selective BRAF inhibitors. Adv Pharmacol 65: 27-43.
- Lai F, Guo ST, Jin L, Jiang CC, Wang CY, et al. (2013) Cotargeting histone deacetylases and oncogenic BRAF synergistically kills human melanoma cells by necrosis independently of RIPK1 and RIPK3. Cell Death Dis 4: e655.
- Study of HBI-8000 With Nivolumab in Melanoma, Renal Cell Carcinoma and Non-Small Cell Lung Cancer.
- Khushalani NI, Brohl AS, Markowitz J, Bazhenova LA, Daniels GA, et al. (2020) Significant anti-tumor activity of HBI-8000, a class I histone deacetylase inhibitor (HDACi) in combination with nivolumab (NIVO) in anti-PD1 therapy-naïve advanced melanoma (TN-Mel). Poster presentation SITC 2020 Annual Meeting.
- Ahuja N, Easwaran H, Baylin SB (2014) Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest 124: 56-63.
- Azad N, Zahnow CA, Rudin CM, Baylin SB (2013) The future of epigenetic therapy in solid tumours-Lessons from the past. Nat Rev Clin Oncol 10: 256–266.
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, et al. (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141: 69-80.
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, et al. (2021) Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J Clin Oncol 40(2):127-137.
- Study comparing investigational drug HBI-8000 Combined with nivolumab vs. nivolumab in patients with advanced melanoma.
