info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map

Received: July 13,2018; Published: August 17,2018
*Corresponding author: Angel Sánchez Lamar, Head of Genetic Toxicology Laboratory, Department of Vegetal Biology, Faculty of Biology, Havana University, Cuba
DOI: 10.26717/BJSTR.2018.08.001601
Among Phyllanthus orbicularis K phytocompounds, 2,4-di-terbutilfenol o el 2,6-di-secbutilfenol are proposed as potent mutagenicity inhibitors produced by hydrogen peroxide, in S. tiphimurium. This DNA protected capacity, against oxidative damage can be explained, theoretically, by several ways. To date, has not been reported neither molecular mechanics as support to the antimutagenic activity of this compounds. In the present work in silico modelation was performed in order to propose a chemistry explanation of the scavenging mechanism versus hydroxyls radicals mediated by these phenols. To carried out the model, a quantum mechanics methodology was employed with: i) DFT calculations, employing B3LYP/6-31G** functional, to the structures of the 2,4-di-terbutilfenol o el 2,6-di-secbutilfenol, and the open shell uB3LYP/6-31G** functional to all proposed radicals, ii) Semi empiric calculations to model the final products obtained in the last stage of free radical reaction. The modelled mechanism proposed a hydrogen abstraction from the hydroxyl group mediated by the radical OH- generating phenoxyl free radicals in the propagation stage. These are eliminated in the termination stage by annihilation with other radicals or self-annihilation, generating both monomeric and dimeric of quinonic and phenolic-derived compounds, as final products in the proposed mechanism.
Keywords: Phyllanthus orbicularis; Antioxidant; Desmutagen; DFT; AM1 semi-empiric model
Abbreviations: DTBP: 2,4-Di-Tert-Butylphenol; DSBP: 2,6-Di-Sec-Butulphenol; ROS: Reactive Oxygen Species; RSE: Radical Stability Energy; BDE: Bonding energy dissociations; EF: Eigenvector Following; SOMO: Single Ocupied Molecular Orbital; HOMO: High occupied molecular orbital
DNA damage in somatic cells is involved in the etiology of chronic degenerative conditions such as cancer, atherosclerosis, autoimmune and neurological diseases [1-4]. The current treatment and prevention strategies against this condition consider the fact that plants contain useful metabolites as DNA protectors [5]. In the specialized literature, the Cuban specie Phyllanthus orbicularis Kunh, have been widely supported as DNA protector [6-10]. The aqueous extract obtained from this plant is an effective antimutagenic and antigenotoxic Phyto derived [11]. Using in vitro bioassays was proved the extract capacity to reduce significantly the DNA damage produced by hydrogen peroxide (H2O2) [12,13]. From the extract, were obtained two highly purified chemical fractionations. Each were able to inhibit the H2O2-induced mutagenesis, in bacterial cells that was treated at the same time with the fractions and peroxide. In these fractions was identified the phenolic compounds 2,4-di-tert-butylphenol (DTBP) and 2,6-di-sec-butulphenol (DSBP) as responsible of inhibition of H2O2-induced mutagenesis [14].
Plant phenolic compounds are the most frequently reported substances, responsible for antimutagenic properties shown to be fine antioxidants and potent modulators of xenobiotic metabolism enzymes, among other activities. The scavenging of reactive oxygen species (ROS) able to produce DNA oxidative damage, is one of the most refereed mechanism in literature to explain the action way for some nature desmutagenic agents [15,16]. Many plant- derived compounds like resveratrol, flavonols, alpha tocopherol, lignophenols, quercetins, curcumins, and others, are able to act as ROS scavengers [17-22]. In regard to DTBP and DSBP, previous experimental results suggest that exert a potent antioxidant effect [23]. Nevertheless, to date there are not experimental studies that explain the molecular mechanism to the antioxidant activity of these two phytocompounds.
The current development of computational chemistry has been established theoretical procedures to help the investigation and the understanding of phenomena of chemical and biological interest using computational techniques. The employ of mathematics algorithm and mechanic quantum methods make possible the study and modulation of several systems, especially those with biochemistry nature [24-26]. Several authors had informed the theoretical modulation of related mechanisms with reactions between phytochemical compounds and ROS [27-29]. The aim of the present work was the study of the molecular mechanism involved in the 2,4-di-tert-butylphenol and 2,6-di-sec-butylphenol antioxidant action against HO• radicals. To do this, we will use the theoretical procedure of computational chemistry, modeling in silico the scavenger reactions against HO• radicals mediated by these phenols.
DFT calculations were performed using the Gaussian 03 package [30] and AM1 semiempiric calculations using the MOPAC 6.0™ program package (MOPAC2009).
Among different combinations of density functional theory methods and basis sets, the B3LYP/6-31G (d,p) method has proven reliable in reproducing the geometries, frequencies, and bond lengths [26]. Hence, that we used this functional to optimize the phenolic molecular geometries used in the current work. The unrestricted open-shell approach uB3LYP and 6-31G (d,p) basis set Here et al.; Clark et al.; Frish et al. were used to optimize the geometry of their radicals and hydroxyl radical. No symmetry constraints were used with the default convergence criteria. To the molecular orbital analysis and their topologies in all calculations was included the key words POP=REGULAR, which give information about the orbitals coefficients. The atomics charge densities analysis was performed using the Mulliken110 methods. The radical stability energy (RSE) is a parameter that allow calculate the radical stability generate in ROS reactions and is calculated by the formula:
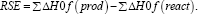
The bonding energy dissociations (BDE) is an intrinsic thermodynamic parameter that define the homolitic dissociation facility in O-H and C-H bonds and generate radicals. The BDEs for the generation of the respective radicals from the parent compounds are calculated by the formula:
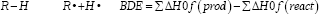
In MOPAC calculations, the geometries of the possible final products from the free radical reactions were optimized and their total energies evaluated using the semiempiric Hamiltonian AM1 method. The eigenvector following (EF) optimization procedure was carried out in all calculations. The precise command was used to avoid some mistake in MOPAC calculations. The command vector was used with the aim to obtain in Keywords formation about the structures electronics properties.
From the OH« radical geometric optimization, was obtained a compound with a lineal geometry, formed by two atoms to a distance of 0,975 A between their cores. The atomic charge densities values obtained for each atom were: +0,247 to the hydrogen and -0,247 to the oxygen. This last element polarized toward itself the electronic cloud in the O-H bond affecting the contributions from the hydrogen atomic orbitals to the SOMO molecular orbital, so oxygen is the atom that most contributes to it with 99.74%. This means a greater probability to find the unpaired electron on oxygen, implying that the covalent bond with another molecule will be formed by the oxygen atom of the radical.
The obtained structures from the phenols geometrical optimization at DFT level are shown in Figures 1 & 2. Table 1 shown the values of atomics densities charges and contributions to high occupied molecular orbital (HOMO), calculated in the phenols functional groups. This allowed define the studied phenols' higher reactivity molecular sites. The atom with the greater negative charge density was the hydroxyl oxygen O (7) in both phenols. Additionally, some carbons in the rings are activating with negative charge density, especially, in para positions 3 and 5 of the two phenols. Hence that the atom with the greater HOMO contribution was the oxygen 7 in the hydroxyl group in both DSBP and DTBP, making this position the most probable to interact with the OH« radical.?
Figure 1: Geometries structures of phenols optimized at DFT level (B3LYP/6-31G**), DTBP (left) and DSBP (right).

Figure 2: Dislocation of the free electron in the resonant structures once is carrying out the hydrogen abstraction: DTBPr-1 and DSBPr-1: in the hydroxyl group; DTBPr-2 and DSBPr-2: in the aromatic ring.

Table 1: Atomics charge densities and contributions to the HOMO (percent values) in the functional groups of the phenols using DFT level (B3LYP/6-31G**).

The interactions between the HO• radical and phenols, respective to the propagation stage in the free radical's reactions, was analyzed as two different possibilities depending of the functional group in the phenolic compounds that suffer the hydrogen abstraction mediated by the HO• radical. The interaction with the hydroxyl group generate the radical named as 1 and, with the aromatic ring, the radical named as 2, in both phenols (DTBPr-1, DSBPr-1, DTBPr-2, DSBPr-2). BDE and RSE calculations were implemented over the resulting phenolic structures to consider the hydrogen abstraction from the atoms O (7) or C (3) for the DTBP and, the O (7) or C (5) for the DSBP, obtaining the radicals DTBPr-1, DTBPr-2, DSBPr-1, and DSBPr-2, respectively. The calculations results obtained are summarize in Table 2.
The previous results allow inferring that is most probable the hydrogen abstraction from the OH group of both phenols. This is because the phenoxyl radicals generated present lowest values of BDE, RSE and relative energy than those generated for the hydrogen abstraction from the aromatic ring. In other hand, based in the Valence Bond (VB) Theory, was obtain that DTBPr-1 and DSBPr-1 present an additional resonant structure to TBPr-2 and DSBPr-2 (Figure 2), that support the results obtained in the modelation. From the obtained values can be inferring that of both most stable radicals, DTBPr-1 and DSBPr-1, the last one is more efficient. The intrinsic thermodynamic parameter BDE to the reactive O-H bond, for the DSBPr-1 was lower than in the DTBPr-1 molecule (Table 2), so this bond will break most easily in the first case. Also, the generated intermediary radical is more stable in DSBP molecule, because this presents a lower value of RSE as shown the calculation (Table 2), so this will be generated in larger quantities. The contributions to the SOMO from the elements that compound the principal functional groups in phenoxyl radicals are shown in Table 3, they are presented just until the atom 10 since the other elements showed a low contribution to this orbital (data not shown). The atoms O (7), C (2), C (4) y C (6), in both phenoxyl radicals, shown the highest contributions to the SOMO, so in that positions is most probable to find the unpaired electron.
Table 2: Thermodynamics parameters obtained from the DFT level calculation (uB3LYP/6-31G**) for the proposed radical of the phenols. E*: relative energy, the lowest value is take as zero and the others values are the difference between their value and the lowest one (this parameter was calculated for each radical independently).

Table 3: Contributions to the SOMO (percent values), of the atomics orbitals from the elements that compound the phenoxyls main functional groups.

The final products in the free radicals' reactions termination stage, were modeled based on the following possibilities:

ArOH + ArO • PP + ArO •
ArO o•+ HO • PT
ArO • + ArO • D
Where, ArOH generically represent both DTBP and DSBP, ArO• is the phenoxyl radical that includes the forms DTBPr-1 y DSBPr-1, PT encompasses all products in the annihilation reactions between the radicals formed from the phenols and the HO•, Pp represent the products obtained from the reactions between the phenoxyls radicals and phenols, D is the possible results dimerization from the interaction of two ArO•. Possibles products derivates from the reaction between ArOH and ArO•. For the DTBPr-1 three possible products were modeled resulting from the hydrogen addition given those atoms with the greater contributions to the SOMO, C (2), C (6) and C (7), assigned as quinonics compounds (Qc2, Qc6 y Qc7 respectively). In other hand, for the DSBPr-1 two possibilities were analyzed, the hydrogen addition in orto (Qo) and para (Qp) positions respect to the oxygen atom give the high symmetry of this molecule. Table 4 shown the values of heat of formation related to the obtaining quinonics products in both phenols.
Table 4: Heat of formation theoretical values of the derived products in the reaction between the phenoxyl radicals and their phenols.

The quinonic structure Qc6 seems to be the most DTBP probable product to obtain, because it presents the lowest heat of formation making it the most stable. The stability of this product may be due to this carbon unlike C (2) and C (4), it's not bonding to bulky tert-butyl groups which induce angular tensions with the hydrogen atom through a steric impediment to be linked to the same carbon, increasing the molecular energy. The products Qc6 and Qc2 present tautomerism in the ceto group and the conversion to the enol form yield the original DTBP ready to start the cycle again. From the DSBP products, the most stable is the quinonic structure formed by the hydrogen additions in para position (Qp). In this molecule the orto positions, like carbons 2 and 4 in DTBP, have linked bulky secbutyl groups and the hydrogen additions in this position generate steric interactions, increasing their energy making less stables like shown the calculations (Table 4).
- ArO•+OH annihilation reaction. The intermediary radical forms DTBPr-1 and DSBPr-1 can react with radicals HO• or self- annihilate. Table 5 shown the theoretical values of heat of formation for the products of termination reactions between the phenoxyl radicals and the hydroxyl radical. The most stable termination product for the DTBP is the one that correspond with the radical OH addition in the non-substituted carbon in position six (C (6)). Again, the steric factor plays a determinant role obtaining this product. The addition of HO• at the substituted positions C (4) and C (2) presents steric impediments due to the bulky substituents at these positions, generating less stables products. In the DSBP products happens something very similar, due to this molecule present bulky substituents in orto positions, the most stable product given their highest stability is the one where the HO« attack the non-substituted carbon in para position.
Table 5: Heat of formation theorica! values of the derived products in the reaction between OH radical and phenoxyls (AM1 semi empiric calculations).

In other hand, the phenoxyl radicals can form dimers by the self-annihilation. For the DTBPr-1 case was analyzed the possibility formation of 10 dimeric complexes. In the DSBPr-1 case just three possibilities were analyzed. The heat of formation theoretical values of all possible tautomeric form was calculated, the results are summarizing in Table 6. The lowest energy structure for the DTBPr-1 dimers was the one formed by the interaction between the C (6) of two radicals (C (6)-C (6)). In the ceto-enolic tautomeric equilibrium was obtained that the dienolic form of C (6)-C (6) is even most stable that the diceto form with a lower AH0f. For the DSBPr-1 dimers, the complex with the lowest energy was the dienolic form obtained from the interaction between the C (4), the same carbon with the highest contribution to the SOMO.
Table 6: Heat of formation theorical values of the derived products in the dimerization reactions (AM1 semi empiric calculations).

The current medicine strategies for anticancer therapy take in count the inclusion of natural products for prevention and chemotherapy of this illness. It is knower that in the etiology of carcinogenic proliferation the somatic DNA damage is involved [31,4]. So, the searching of new active molecules from vegetal origin, able to reduce the cancer risk, is a scientific task of high significance for the sustenance of human health [1]. In this context, the 2,4-di- tert-butylphenol and 2,6-di-sec-butylphenol have been proposed as active phenols molecule able to protect DNA against oxidative- damage induced by HO« radicals [14]. Regarding these two compounds, the present work constitutes the first computational study about the molecular mechanism involved in its antioxidant action. This in silico chemical study starts from the previously reported comprehension about the interactions between free radicals and phenolic compounds [29]. This interaction type can be produced by the radical addition to the aromatic ring, or by hydrogen abstraction. The occurrence of one or another mechanism is determinate fundamentally by the stability of the radical that are formed. In the last case there are two possibilities of abstracting hydrogen: from a hydroxyl group, or from the aromatic ring [32].
The HO« radical is electrophilic, these generally tend to react easily with phenolic compounds through the hydrogen abstraction mechanism [33-36]. In these reactions, the radical Single Ocupied Molecular Orbital (SOMO) and the phenolic SOMO frontier orbitals are involved [37]. The geometric optimization analysis of this work permitted the identification of the principal reactive atomic domains in the molecular structures of each interacting chemical species. The obtained result about OH« radical's geometric optimization suggested that the covalent bond with another molecule will be formed by the oxygen atom of the radical. This is accord with the reported by other authors about optimization of this molecule [3841]. In the case of the molecular structures of both phenols, the positions most likely to be attacked by the HO « radical were the hydroxyl group and the aromatic ring.
The first interaction phase generated the radical forms DTBPr-1 and DTBPr-2, from DTBP molecule and, DSBPr-1 and DSBPr-2, from DSBP molecule. At the same time, this reaction yields water of the hydroxyl radical, which constitutes a way of reducing the concentration of HO « radical in the internal cellular environment. The interactions between HO« radical and phenols, respective to the propagation stage of the free radical's reactions, was characterized by the formation of several resonant structure due to dislocation of the free electron, in dependent of the atomic positions of phenols where was carrying out the hydrogen abstraction. These generated phenoxyl radicals remain in the system and have the possibility of continuing to interact with the other molecular species of the radical reaction system. In the termination stage of free radical reactions, two main types of molecular interactions were modeled: i) the phenoxyl radicals with their respective original phenolic forms, and ii) the annihilation reactions between existing radicals in the reaction system. Among these last ones, the attack by the HO« to the positions of the phenolic molecules with unsubstituted carbon was a common characteristic to the annihilation reactions produced between DTBPr-1 or DSBPr-1 with the radicals HO«.
This is accord with the experimental results obtained by other authors, who measured the spin population in the phenoxyl radical, and the carbon in para position resulted be the most enriched in electronic cloud [42,43].
The set of obtained results allow elaborating theoretical outline ofthe global process (Figure 3). In terms of molecular events for DNA protection is possibel to said that, inside cells the HO• radicals are inhibited, before they reach the genetic material, reacting quickly with phenols (ArOH) generating the phenoxyls radicals (ArO•). The ArO• react then whit ArOH to form quinonic structures that tautomerize to enolic form regenerating ArOH. This event makes a recurrent cycle of reactions where, the OH radicals are inactivated while they are in a cellular medium, and the ArO• are accumulated in the system.Is important to point that, the ArO• radicals replacing the OH have high values of energy in the SOMO so they are less electrophilics, also they hold higher molecular mass. Hence, we can infer that the phenoxyls radical are less reactive and present a lower diffusion coefficient in the cellular inside, consequently they are less toxics for the DNA. In addition, the generated products in the proposed reactions here are structures that have not been recognized to date as toxics for the DNA.
Figure 3: Outline of the global process of patterned radical reactions.

Coincidentally, the dimer proposed here as final product for the DTBP, using in silic methods, was experimentally found in the same chemical fraction from which this phenol was isolated and purified [23]. A possible explanation to the dimer presence in the aqueous extract of P. orbicullaris, is that the proposed mechanism here is taking place as a defense way against the oxidative stress that this plant is underwent in their environment. From the experimental point of view, future chemical or biochemical assays will confirm the formation of the final products proposed by the reaction system modeled in this work. In conclusion, the chemical model here proposed, cover a mechanism of reaction that capture hydroxyl radicals generating the phenoxyl radicals whose self-annihilate obtaining as products monomerics or dimerics quinonics structures. In the cellular environment this means that the generated free radicals from the hydrogen peroxide spontaneous decomposition are consumed, decreasing the rate of DNA damage by two different ways.
The reaction between the hydroxyl radical and phenols, yielding the phenoxyl radicals and water constitute a first way in the HO• concentration reduction. In other hand, the direct annihilation reactions between the radical phenols forms and the HO«, constitute a second way. In addition, the regeneration of the original phenolic forms involves a cyclic reaction system where HO« radicals are inactivated continually while they are present in the cell medium. This justifies the genoprotection that these phenols exert against oxidative stress. Finally, this theoretical model supports the classifying of DSBP and DTBP compounds as desmutagenic agents in the DNA protection.
This work was supported by the Ministery ofScience, Technology and Environment of Cuba (Project No. P223LH001-047 Associated to National Program of Basic Sciences). The authors appreciate the possibility to run the DFT and Semiempiric calculation programs on computer system installed at INSTEC of Havana University


























