 Case Report
Case ReportAbstract
Diagnosis of Bardet-Biedl syndrome (BBS) that distributes worldwide is still dependent on a careful observation of clinical manifestations in spite of enormous progress in genetic testing. Reports on clinical phenotypes of BBS from the Far East are limited. A 33-year-old Japanese man fulfilling diagnostic criteria for BBS showed unusual clinical feature. Firstly, he had motor-dominant peripheral neuropathy in the absence of pathologic variant in peripheral myelin protein 22 gene (PMP22). Secondly, he lacked retinal dystrophy, which is very atypical for a BBS patient in the fourth decade of life. To our knowledge, the above constellation of clinical features is previously unreported in BBS. Whether the patient represents a rare case, or one specific to a Far East subgroup remains unknown. The diagnosis and outcomes of BBS patients may be improved by searching out similar patients.
Keywords: Bardet-Biedl syndrome; Peripheral neuropathy; Retinal dystrophye
Abbreviations: DM: Diabetes Mellitus; FSH: Follicle-Stimulating Hormone; LH: Luteinizing Hormone; CKD: Chronic Kidney Disease; BBS: Bardet-Biedl Syndrome
Introduction
Bardet-Biedl syndrome (BBS; OMIM #209900) is a pleiotropic genetic disorder, or ciliopathy, with autosomal recessive inheritance [1]. Although 19 to 21 BBS genes have been identified to date [2,3], interfamilial and intrafamilial variations in clinical phenotype are not fully reconciled by interactions between BBS genes [4]. In 20% of BBS families, mutations of known genes are not evident [5,6]. Diagnosis of BBS is heavily reliant observing clinical features, rather than genetic analysis [6-9]. Here, we report a 33-year-old Japanese man who was diagnosed with BBS in the first decade of life. He showed, in the fourth decade of life, an unusual combination of symptoms. He lacked possibly the most important BBS feature; retinal dystrophy. Furthermore, he exhibited motor-dominant peripheral neuropathy, which to
Case Report
A 33-year old Japanese man was transferred to the Division of Nephrology because of end-stage renal disease requiring hemodialysis. His was followed-up for 5-years as an outpatient at the Division of Neurology and Endocrinology in our institute. According to the past medical records, he was born normally at term to double first cousin parents with uneventful prenatal periods. At three months of age, he underwent initial surgery for postaxial polysyndactyly. He started walking by himself at the age of 12 months. He had mental and growth retardations in childhood. His skeletal ages at the chronological ages of 3.5 and 12.25 years were 1.25 and 8.5 years, respectively. At 8 years of age, he underwent surgery for unilateral cryptorchidism. His parents stated he was obese and had round face in his childhood. He had gone through a total of four surgical operations for polysyndactyly. An X-ray photograph of the hands taken in the fourth decade of life clearly shows an abnormality of the DIP joint and phalanges (ossa digitorum manus) of the first finger (Figure 1). At 15 years of age, he was diagnosed with type 2 diabetes mellitus (DM): his height, weight and IQ were 143.2 cm (-3.8 SD), 39.0 kg (-1.8 SD) and 66, respectively. Although his blood sugar control was poor, his behavioral problems had prevented him from receiving insulin therapy until he came to our institute at the age of 28 years. Fundoscopic examination at the Department of Ophthalmology revealed the presence of diabetic retinopathy and the absence of retinal dystrophy (Figure 2). In the same year, he visited the Division of Neurology where he was clinically diagnosed with motor dominant peripheral neuropathy. Sensory neuropathy was minimal at this time. Serum testosterone level was within normal range, although serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) were slightly elevated.
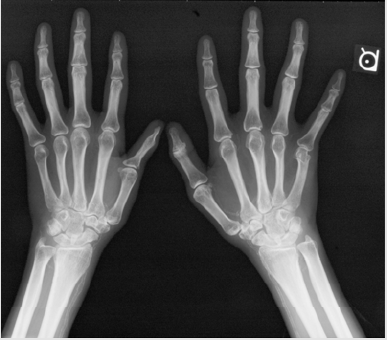
Figure 1: Abnormal X-ray photograph of the hands showing changes to polysyndactyly post-operatively (A). Pronounced arterial calcification is evident (A). CT scan of the abdomen. Kidney size and shape are intact. Note the absence of aortic calcification (B).
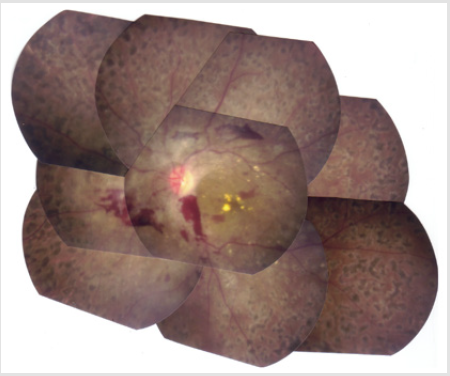
Figure 2: Fundus shows diabetic changes, however there were no indications of retinal dystrophy.
He had stage 5 chronic kidney disease (CKD), when he visited our division. CT imaging failed to show severe cortical atrophy, despite being at stage 5 CKD (data not shown). His restlessness, and a lack of cooperation prevented us from performing percutaneous renal biopsy. Calcification of deferent duct was present (data not shown). The size of external genitalia was normal (data not shown). Neurological examinations of the patient for the past five years have revealed mental retardation to a moderate degree and slowly-progressive atrophy of the distal lower extremities and feet. Although both sensory and motor functions were affected, motor nerve impairment was much more prominent than sensory nerve impairment. Pes cavus (Figure 3) and delay in peripheral nerve conduction velocity (data not shown) were seen. DNA-sequence analysis showed that peripheral myelin protein 22 (PMP22), a gene responsible for HNPP (Aarskog and Vedeler [2000]), was not mutated (data not seen). Another fundoscopic examination was performed at the age of 33 years. His vision results were; Vd=0.5(0.6x + 0.5 Cyl-0.5Ax20), Vs=0.4(0.6x + 1.5 Cyl-1.0Ax170. These results, together with electroretinographic findings (data not shown), further confirmed the absence of retinal dystrophy in his fourth decade.
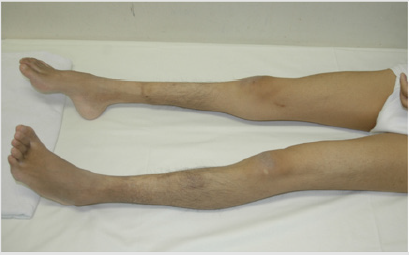
Figure 3: Pes cavus and Champagne bottle appearance of the right leg.
Family History
The patient’s parents are double first cousins who came from Yamanashi prefecture in Japan. He has an elder brother who has no BBS phenotypes. His maternal grandmother and grandfather had diabetes mellitus and cerebral infarct, respectively. His paternal grandmother had cardiovascular disease (details unknown) and had brothers who were short of stature (140-150 cm).
Discussion
A BBS patient requires 4 primary features, or 3 primary features plus at least 2 secondary features [2,9]: Our patient has at least 3, and possibly 4, primary features; polydactyly, renal abnormalities, learning disability, and obesity. We are not sure if latent hypogonadism and calcification of deferent duct that he has should qualify as another primary feature; “hypogonadism in males”. Nevertheless, it is fair to claim he is diagnosed with BBS, because he has at least 2 secondary features; diabetes mellitus and developmental delay [2,9]. Particular emphasis is placed on eye phenotype in diagnosis of BBS [10]. Retinal dystrophy, rod-cone dystrophy, or related eye disorders are present in 100% of patients in some reports [7,11-13]. Although it was present in slightly less than 100% in other reports, those who did not have the eye disorder were children [8,9]. In one Chinese report, retinitis pigmentosa was found only in 72.3% of the patients [14]. This lower incidence may have been due to a young mean age of 15.8 years in study cohort. Typically, BBS patients develop ocular problems in the second and third decade of life [15]. Our patient is over 30 years of age, and it is very unusual for BBS patients of this age not to have any of the above-mentioned eye phenopypes. BBS genes encode cilium proteins, of which there are reported links with retinal dystrophy [2,5].
Rod-cone dystrophy may be caused by trafficking abnormalities across the defective modified cilia that connects to the inner and outer segments of photoreceptors leading to apoptosis [16]. Clinically, we ruled out diagnosis of Alstrom syndrome (MIM 203800), Prader-Willi syndrome (MIM 176270), Carpenter syndrome (MIM 201000), Biemond syndrome II (MIM 210350), and Cohen syndrome (MIM 216550) based on the patient’s signs and symptoms. It seems justified to state that our patient represents a spectrum of BBS. Genetic analysis might help clarifying this issue, however, parental informed consent was not obtained.<.
Motor-dominant peripheral neuropathy in our patient was already prominent 5 years prior to transfer to the Nephrology Division (data not shown). According to Beales et al., ataxia with poor coordination and dysdiadochokinesia were frequent neurological findings in BBS. None of the 109 patients in that study showed motor-dominant peripheral neuropathy [9]. Sensory dominant peripheral neuropathy is much more common in DM [17]. There are 2 groups BBS patients with renal failure, the first one includes patients with renal and urinary tract abnormalities diagnosed early in life, and the second one includes adult BBS patients with no clear information on the onset of CKD (18). Our patient belongs to the second group. End-stage renal disease due to diabetic nephropathy has the following features [19]: more than 10-year-history of DM, proliferative retinopathy, massive proteinuria, absence of severe cortical atrophy of the kidney. Although our patient had some of these, detailed mechanisms leading to renal failure remained to be elucidated without renal biopsy. BBS patients are found worldwide. BBS phenotype in the Far East remains largely unknown, although the genetic backgrounds of Japanese BBS patients are at least partly consistent with those of BBS patients in other countries [20]. There are case reports [21-24]. In conclusion, constellation of clinical features shown in our patient is previously unreported in BBS. Whether it represents a subgroup in the Far East remains to be seen. Finding patients with similar clinical phenotype may improve the diagnosis and outcomes of BBS patients.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgement
A written informed consent was obtained from this patient’s parents. We thank Prof. Seiji Suzuki, and President Tetsuo Nishikawa, from the Division of Endocrinology, Department of Medicine, Showa University Fujigaoka Hospital, and Yokohama Rosai Hospital, respectively, for giving us clinical information.
References
- Hildebrandt F, Benzing T, Katsanis N (2011) Ciliopathies. Engl J Med 364(16): 1533-1543.
- Khan S, Muhammad N, Khan MA, Kamal A, Rehman ZU, et al. (2016) Genetics of human Bardet-Biedl syndrome,an updates. Clin Genet 90(1): 3-15.
- Heon E, Kim G, Qin S, Garrison JE, Tavares E, et al. (2016) Mutations inC8ORF37 cause Bardet Biedl syndrome (BBS21). Hum Mol Genet 25(11): 2283-2294.
- 4. Badano JL, Leitch CC, Ansley SJ, May-Simera H, Lawson S, et al. (2006) Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 439(7074): 326-330.
- 5. Stoetzel C, Muller J, Laurier V, Davis EE, Zaghloul NA, et al. (2007) Identification of a novel BBS gene (BBS 12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet 80(1): 1-11.
- Forsythe E, Beales PL (2013) Bardet-Biedl syndrome. Eur J Hum Genet 21(1): 8-13.
- Green JS, Parfrey PS, Harnet JD, Farid NR, Cramer BC, et al. (1989) The cardinal manifestation of Bardet-Biedl syndrome, a form of Laurence-Moon-Bardet-Biedl syndrome. N Engl J Med 321(15): 1002-1009.
- Beales PL, Warner AM, Hitman GA, Thakker R, Flinter FA (1997) Bardet-Biedl syndrome: a molecular and phenotypic study of 18 families. J Med Genet 34(2): 92-98.
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA (1999) New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet36(6): 437-446.
- David A, Bitoun P, Lacombe D, Lambert JC, Nivelon A, et al. (1999) Hydrometrocolpos and polydactyly. A common neonatal presentation of Bardet-Biedl and McKusick Kaufman syndrome. J Med Genet 36(8): 599-603.
- Fulton AB, Hansen RM, Glynn RJ (1993) Natural course of visual functions in the Bardet-Biedl syndrome. Arch Ophthalmol 111(11): 1500-1506.
- Riise R, Andréasson S, Brogstrom MK, Wright AF, Tommerup N, et al. (1997) Intrafamilial variation of the phenotype in Bardet-Biedl syndrome. Br J Ophthalmol 81(5): 378-385.
- Young TL, Penney L, Woods MO, Parfrey PS, Green JS, et al. (1999) A fifth locus for Bardet-Biedl syndrome maps to chromosome 2q31. Am J Hum Genet 64(3): 900-904.
- Lian-Jun W, Xueying P, Cunyou D, Xuangying P (1998) Bardet-Biedl syndrome: a review of Chinese literature and a report of two cases. Ophthalmic Genetics 19(2): 107-109.
- Fialkow PJ (1983) Noonan syndrome and other disorders involving multiple organ systems. In: Harrison’s Principles of Internal Medicine. Tenth edition. Edited by RG Peterdorf, RD Adams, E Braunwald, KJ Isselbacher, JB Martin, JD Wilson. New York, USA, pp. 578-580.
- Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, et al. (2004) Bbs2-null mice have neurosensory deficits, adefect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci USA 101(47): 16588-16593.
- Vinic A, Mitchell B (1988) Clinical aspects of diabetic neuropathies. Diabetes Metabo Rev 4(3): 223-253.
- Zacchia M, Di Iorio V, Trepiccione F, Caterino M, Capasso G (2017) The Kidney in Bardet-Biedl Syndrome: Possible Pathogenesis of Urine Concentrating Defect. Kidney Dis (Basel) 3(2): 57-65.
- Darving HH, Mauer M, Ritz E (2007) Brenner & Rector’s The Kidney. In: Brenner BM, Levine SA (Eds.), Saunders, Philadelphia, USA, pp. 1777-1818.
- Hirano M, Satake W, Ihara K, Tsuge I, Kondo S, et al. (2015) The First Nationwide Survey and Genetic Analyses of Bardet-BiedlSyndrome in Japan. PLoS ONE 10(9): e0136317.
- Wolf B (1991) Bardet-Biedl syndrome in Zimbabwean child. Cent Afr J Med 37(10): 341-342.
- Kalangu K (1995) A further case of Bardet-Biedl syndrome. Cent Afr J Med 41(5): 167-169.
- Tonomura Y, Hirano M, Shimada K, Asai H, Ikeda M, et al. (2009) Treatable fluctuating mental impairment in a patient with Bardet-Biedl syndrome. Clin Neurol Neurosurg 111(1): 102-104.
- Ohto T, Enokizono T, Tanaka R, Tanaka M, Suzuki H, et al. (2017) A novel BBS10 mutation identified in a patient with Bardet-Biedl syndrome with a violent emotional outbreak. Hum Genome Var 4: 17033.
