 Research Article
Research ArticleAbstract
Background: Cerebral ischemic disorders can be divided into global and focal. In condition of simultaneous and similar drop of cerebral blood flow global ischemia occurs while whenever there is a partial brain involvement it is defined as focal ischemia. Lots of paper deal with focal ischemia and the relative changes in metabolism or blood supply while few reports deal with global ischemia secondary to the difficulties in creating a representative model.
Objectives: This paper aim to analyze changes in brain metabolism during ischemia in order to achieve a better cognition of the time limit for reversible global ischemia.
Methods: Sixteen guinea pigs underwent global reversible cerebral ischemia. Four of them for 30 minutes and other four for one hour. During the entire procedure pH, pO2 and pCO2 values were recorded while brain metabolites were collected through microdialysis. Secondarily, other 8 pigs were pretreated with L-NAME and monitored as the previous eight to analyze changes in metabolism.
Results: In animals not pretreated with L-NAME anaerobic glycolysis takes place with glucose consumption, increase in pyruvate and lactate levels and pH decrease. L-NAME pretreatment modify those data transforming the anaerobic glycolysis in a partial aerobic one in which glucose decrease is followed by pyruvate increase with stability of lactate and pH levels.
Conclusion: Brain metabolism seems to remain active even after cortical activity stops. Such a finding becomes crucial in defining scientific criteria for brain death and in the research for neuroprotective measures against brain ischemia. Brain microdialysis represents a very accurate monitoring system for neurological Intensive Care Unit (ICU) patients. Pharmacological pretreatment can modify brain metabolism during tamponade demonstrating the persistence of cellular activity even in conditions of supposedly brain death like 30 and 60 minutes of cerebral ischemia.
Keywords: Ischemia; Metabolism; L-NAME; Reperfusion
Abbreviations: ICU: Intensive Care Unit; CBF: Cerebral Blood Flow; ATP: Adenosine Triphosphate; PDH: Pyruvate Dehydrogenate; eNOS: endothelial Nitric Oxide Synthase; nNOS: neuronal Nitric Oxide Synthase; NO: Nitric Oxide; SSS: Superior Sagittal Sinus; SAP: Systemic Arterial Pressure; ICP: Intracranial Pressure
Introduction
Global reversible cerebral ischemia results from transient low Cerebral Blood Flow (CBF) or severe hypoxia and reduction of energy metabolism. Phosphonate compounds, partic ularly Adenosine Triphosphate (ATP) represent brain energy source. Under normoxic condition, more than 95% of ATP is generated by glycolysis and mitochondrial oxidative phosphorylation. For this very reason, most of the mechanism involved in cell death and brain ischemia have mitochondria as a primary target or as a regulator of the mechanisms itself. Hypoxia/ischemia interferes with mitochondrial oxidation of pyruvate leading to cytosolic lactate formation. Such a shift leads to ATP generation only through glycolysis causing a progressive pH decrease. Acidosis development results then in cellular ion homeostasis progressive breakdown. This cascade is on the other hand reversible as in cases of short time ischemia ion gradients gradually normalize with a recovery in metabolism after about 15 minutes. However, due to the inhibition of Pyruvate Dehydrogenate (PDH) complex by free radicals, lactate/pyruvate ratio is still significantly higher than normal. This finding is related to the lack of pyruvate metabolism in the mitochondria. Then, during reperfusion, cell death results from a progressive inactivation and structural damage [1]. The loss in ATP modifies intracellular homeostasis blocking ATP-dependent pumps in favor of calcium dependent ones with an increase in calcium levels that activates endothelial Nitric Oxide Synthase (eNOS) and neuronal Nitric Oxide Synthase (nNOS). Nitric Oxide (NO) levels then rise. competing with O2 during oxidative phosphorylation and subsequently decreasing even more cellular ATP formation [2].
Nonetheless, the timing of the aforementioned metabolic cascades is yet to be clearly defined but, as shown in animal models, reversible global ischemia can lead either to deterioration of the neurological status either to recovery [3,4]. Furthermore, there are no clear evidence throughout the literature on how long global cerebral ischemia has to last to produce irreversible neuronal damages in humans. Many reports instead suggest how specific neuronal populations vary substantially in ischemic tolerance [5,6] and how this tolerance can be modified by chemical and physical agents [3,7]. The still open questions mentioned before represent one of the major issues in clinical practice. In fact, guessing the severity of brain damage and the correct timing for treatment is vital in the choice whether to treat or not and in the choice for the proper neuroprotective measures. In order to clarify those questions in our paper an evaluation of brain metabolism during tamponade was carried on in basal conditions and after Nitro-Larginine methyl ester (L-NAME) administration. L-NAME was chosen for its interference with NO synthesis being such a molecule strongly involved in brain metabolism during ischemia.
Material and Methods
The experimental protocol followed the Guidelines for the Care and Use of Laboratory Animals of the Ethical Committee of our Institution or the Helsinki Declaration of 1975 (and as revised in 1983) and the Gunma University Graduate School of Medicine experimental protocol [8]. Eight adult guinea pigs weighting 25- 30 g each underwent global reversible ischemia that was induced using a Fogarty balloon inflated intracranially. In four cases brain tamponade lasted for 30 minutes while in other four it lasted 60 minutes. Blood specimen were collected from the Superior Sagittal Sinus (SSS), the right femoral artery and the right femoral vein in order to measure pH, pO2 and pCO2. Cerebral microdialysis was performed to analyze brain metabolites (glucose, pyruvate, lactate, glycerol). A second experiment was performed on other eight animals that were submitted to brain tamponade similarly to the previous ones adding L-NAME pretreatment.
Surgical Preparation
Specimens were placed in sphinx position on the operating table. Anesthesia was induced (Atropine 0,5-1 mg Ketamine hydrochloride 10 mg/Kg, Halothane 0,8-1%, Pancuronium Bromide 4 mg), and then maintained (Pancuronium Bromide 0,5 mg/h, artificial ventilation with O2 40% and Halothane 1% mixture). Right femoral artery was cannulated with a 20G peripheral venous catheter to obtain Systemic Arterial Pressure (SAP) and arterial blood specimen. The animals were maintained in normotensive and normocapnic states, during anesthesia. Rectal temperature was maintained at 38°C using a heating blanket.
A wide skin, fascia and muscle incision was performed to
expose the skull. The listed five different surgical procedures were
performed on each specimen.
a) Left temporal burr hole to monitor Cerebral Blood
Flow (CBF) velocity through middle cerebral artery (MCA)
ultrasound Doppler
b) Left frontal 3x2 cm mini-craniotomy 3 mm to the left from
the sagittal line and a 1,5 cm anterior to the coronary suture for
Intracranial Pressure (ICP) monitoring. After opening the dura,
a piezoelectric intraparenchymal ICP Codman ® Microsensor
(CODMAN ® ICP EXPRESS Monitor system, Codman and
Shurtleff, Inc., MA, USA) was placed to monito ICP
c) Burr hole on the sagittal line 5 mm anterior to the
coronary suture to place a 20G Tuohy needle to get venous
blood specimen from the SSS
d) Right frontal mini-craniotomy 5 mm right from the sagittal
line and 1,5 cm anterior to the coronary suture to insert a CMA
70 brain MD microdialysis catheter ® (CMA microdialysis AB,
Soina, Sweden) into the subdural space after opening the dura
e) Burr hole was made on the right occipital bone 5 mm
from the midline and 1,5 cm posterior to the coronary suture to
insert a Fogarty catheter (20 ml max. capacity).
All animals’ cortical activity was monitored with scalp
multichannel Electroencephalography (EEG). During the
monitoring time no mortality occurred, and, at the end of the
observational time, the animals were sacrificed according to the
guidelines reported in the previous paragraph. As the focus of the
paper was the metabolic change in brain parenchyma no clinical
evaluation was performed.
Evaluation of Brain Metabolism
Thirty minutes after anesthesia induction, brain tamponade was obtained inflating the Fogarty catheter with Saline Solution 0,9% till observing reverberant wave at the Doppler ultrasonography and null CBF. Doppler ultrasound was used only to demonstrate the achieving of absent blood flow. For this very reason no ulterior measure of CBF were taken. Brain reperfusion was obtained deflating the Fogarty catheter until recording again CBF and witnessing ICP decrease. Blood specimens were collected from the SSS, right femoral artery and vein while brain metabolites were extracted trough microdialysis(sampling of the interstitial fluid via a double-lumen probe with an integrated semipermeable membrane infusing constantly lactate-free Ringer’s solution at a fixed flow rate of 2 mcg/min [9]).
In four animals (A30, B30, C30, D30) brain tamponade lasted 30 minutes obtaining samples at the following time intervals: T0=baseline; T1=start brain tamponade; T2=start brain reperfusion after 30 minutes of brain tamponade; T3= first 30 min of brain reperfusion; T4=60 min of brain reperfusion; T5=90 min of brain reperfusion; T6=120 min of brain reperfusion and T7=150 min of brain reperfusion. Repeated measurements were taken to have a proper evaluation of the metabolic changes alongside the whole experiment in order not to miss unexpected values modifications. Other four animals (A60, B60, C60, D60experienced 60 minutes of brain tamponade and were evaluated similarly to the previous ones. Time intervals were: T0=baseline; T1=start brain tamponade; T2=first 30 minutes of brain tamponade; T3=start reperfusion after 60 min of brain tamponade; T4=30 min of brain reperfusion; T5=60 min of brain reperfusion; T6=90 min of brain reperfusion, T7=120 min of brain reperfusion and T8=150 min of brain reperfusion. To further evaluate the changes in brain metabolism during ischemic time another group of eight animals (A30+, B30+, C30+, D30+, A60+, B60+, C60+ and D60+) was tested adding L-NAME pretreatment. L-NAME is an antagonist of the eNOS and nNOS [10] which reduce NO levels promoting a switch in energetic metabolism to creatinine-phosphate/creatinine system (CrP/Cr) [11,12]. L-NAME was administered intravenously at the dosage of 1 mg/Kg [13,14]. No measurement of L-NAME metabolites was undertaken as this was not the scope of the paper
Results
30 Minutes Brain Tamponade
Complete data for A30, B30, C30 and D30 are reported in Supplemental material I. Mean brain baseline ICP was 15 mmHg reaching arterial pressure-like values during brain tamponade and dropping back to normality during reperfusion. SAP decreased only during brain tamponade. EEG monitoring during tamponade confirmed the absence of cortical activity. SSS venous baseline mean pH value was 7,449 while, after tamponade, it decreased to 7,140 tending to recover after 150 from reperfusion reaching 7,420. SSS PaCO2 and Pa O2 baseline values were 37,7 and 51,5 mmHg respectively. These values showed the same trend during tamponade with a maximum increase of PaCO2 and decrease of PaO2 at the end of the 30 minutes (PaCO2=82,9 mmHg and PaO2=44,5 mmHg) tending to normalize at reperfusion (Table 1).
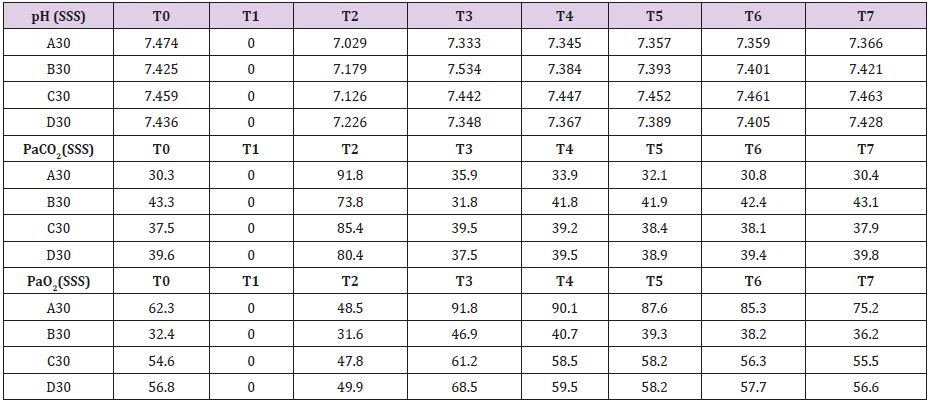
Table 1: SSS value in basal condition.
Mean baseline glucose value was 0,886 mmol/L. It decreased during ischemia to 0,725 mmol/L, decreasing even further after 30 minutes from reperfusion (0,174 mmol/L). Those values recovered and reached higher levels than baseline only after 60minute from reperfusion. Lactate increase dramatically during brain tamponade reaching the maximum level 60 minutes after reperfusion (10,084 vs 2,737 mmol/L) tending afterword to return to baseline. Glycerol constantly increased until 60 minutes after reperfusion. Those values were maintained higher than baseline even later on. Mean baseline pyruvate value was 147,8 μmol/L. This value slightly increase after 30 minutes tamponade (164,3 μmol/L), reaching its lowest after 30 minutes from reperfusion (83,8 μmol/L) and its highest 60 minutes after reperfusion (421,1 μmol/L). Lactate/ pyruvate ratio tend to remain constant at the end of brain tamponade (20,8 vs 18,5) while there is a tendency to increase after 30 minutes from reperfusion (89,0).
60 Minutes Brain Tamponade
In Supplemental material II are reported the results for the 60 minutes brain tamponade animals (A60, B60, C60, D60). CBF, ICP and SAP showed a similar trend to what seen in the previous experiment while EEG activity differs significantly. In fact, while during tamponade the EEG disappeared as in the previous experiment, after reperfusion there was no recover. SSS venous pH remained stable for the whole experiment (7,500) except at the end of the 60 minutes when it reached 7,109. SSS PaCO2 at the baseline was 32,7 mmHg increasing constantly to 111,5 mmHg at the end of brain tamponade. Such increase was reverted to baseline at the end of reperfusion. SSS PaO2 increased steadily from baseline values of 32,7 mmHg till reaching its maximum at the end of brain tamponade. Return to baseline is achieved after 60 minutes from reperfusion (Table 2). Mean glucose value (0,886 mmol/L at baseline) reached almost 0 mmol/L after 60 minutes of brain ischemia. Only after 90 minutes of reperfusion such a level started to grow again reaching baseline after 120 minutes. Lactate levels increased up to 8 times initial values while glycerol ones reached levels 5 times higher than baseline. Pyruvate levels increased to 170,4 μmol/L (baseline of 68,8μmol/L) decreasing at the end of brain tamponade reaching almost 0 μmol/L 30 minutes after reperfusion. Finally, lactate/pyruvate ratio increase progressively until 30 minutes after reperfusion maintaining such higher values for the whole experiment.
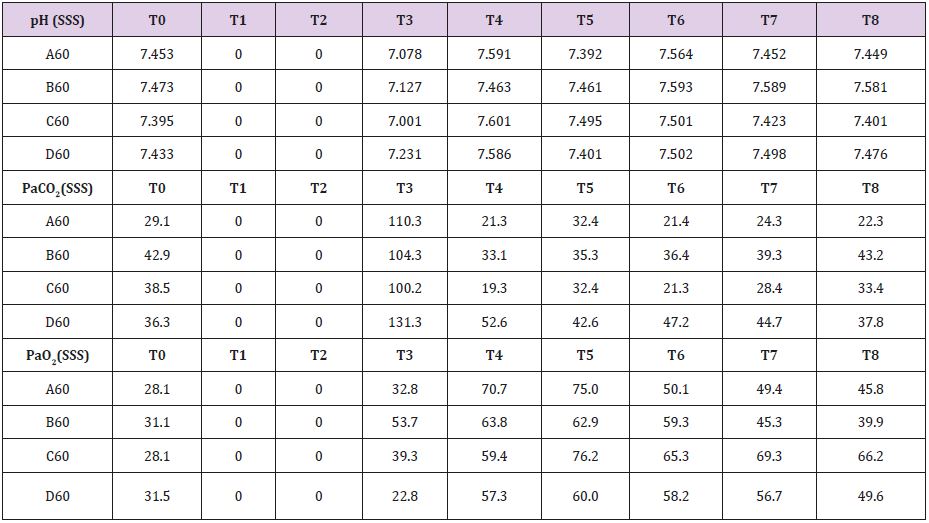
Table 2: SSS value in basal condition.
30 Minutes Brain Tamponade after L-NAME Pretreatment
In Supplemental material III are reported the measured values for the four animals pretreat with L-NAME. SSS venous baseline mean pH value was 7,475; this value remained quite unchanged after 30 minutes of brain tamponade reaching 7,492. After 150 minutes from reperfusion pH value reached 7,518. SSS PaCO2 and PaO2 baseline values were 39,15 and 51,1 mmHg respectively. These values showed the same trend during tamponade while differing from the one measured without L-NAME pretreatment in their trend (PaCO2=30,6 and PaO2=63,9 mmHg) (Table 3). Baseline mean glucose value was 1,625 mmol/L. During tamponade glucose levels decreased till 0,137 mmol/L. After reperfusion glucose levels rose up to 1,761 mmol/L at 150 minutes from it. Lactate levels follow a different tendency during brain tamponade with L-NAME pretreatment (26,3 vs 17,87 mmol/L at baseline); in fact, the measured increase is much subtler and more gradual compared with non-treated animals. Glycerol constantly increased even after 150 minutes from reperfusion. Mean baseline pyruvate value was 226,5 μmol/L. This value slightly increase after 30 minutes tamponade (165,5 μmol/L), reaching its lowest after 30 minutes from reperfusion (106,2 μmol/L) to increase again progressively up to 248,5 μmol/L after 150 minutes from reperfusion. Finally, lactate/pyruvate ratio tends to remain constant.
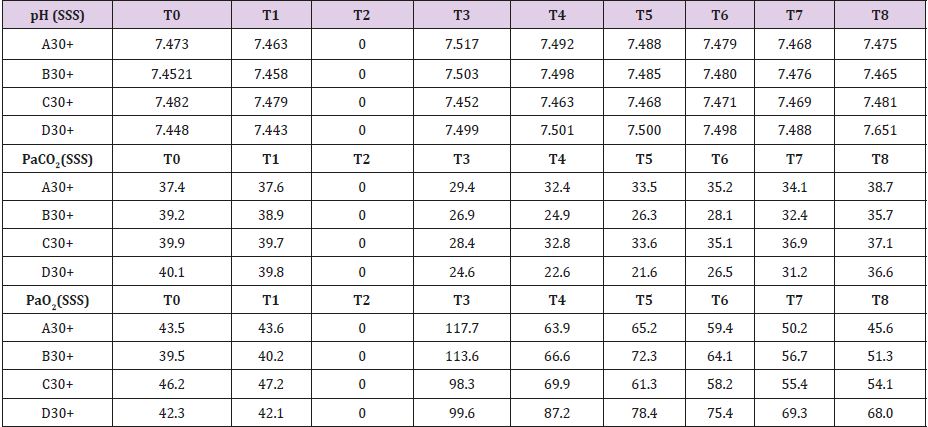
Table 3: SSS value after L-NAME administration.
60 Minutes Brain Tamponade After L-NAME Pretreatment
Measured values are reported in Supplemental material IV. SSS venous baseline mean pH value was 7,468. It remains quite stable during the whole process. Similar tendencies are shown by PaCO2 and PaO2 (Table 4). Baseline mean glucose value that was 1,387 mmol/L almost reached 0 mmol/L after 30 minutes of brain ischemia returning to baseline only after 150 minutes from reperfusion. While lactate and pyruvate levels remained stable glycerol, ones reached levels 10 times higher than baselines.
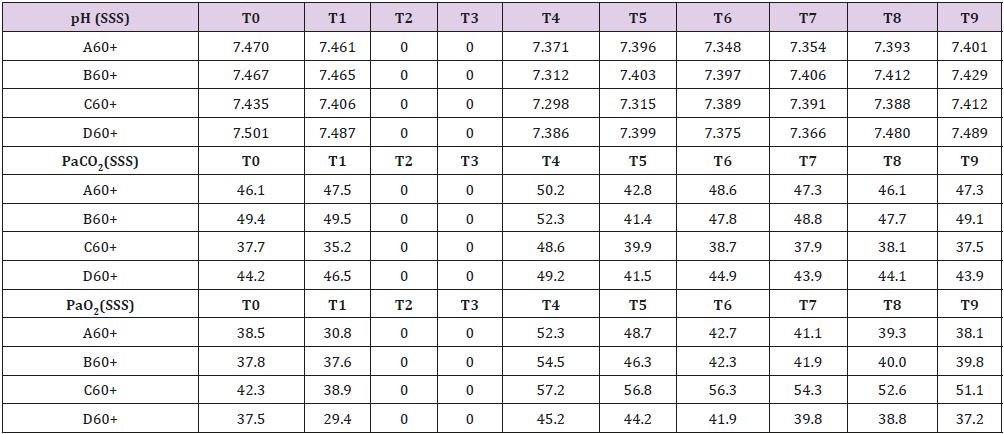
Table 4: SSS value after L-NAME administration.
Statistical Analysis
Secondary to the difficulties in creating an animal model for prolonged global cerebral ischemia only 16 guinea pigs were collected divided, as mentioned before in 4 groups of 4 samples. For this reason, having a statistically significant analysis was not possible being de facto a limit of our work that had to remain observational and inferring on the results. We are well aware of such a limit thus suggesting further studies to invigorate our results. Nonetheless, mean and standard deviation for samples collected from the SSS were calculated and reported graphically in Figure 1 and Tables 5 & 6. Those were the chosen value studied being the most representative for post tamponade metabolism. From the graph a tendency in improvement of metabolism after L-NAME pretreatment can be guessed.
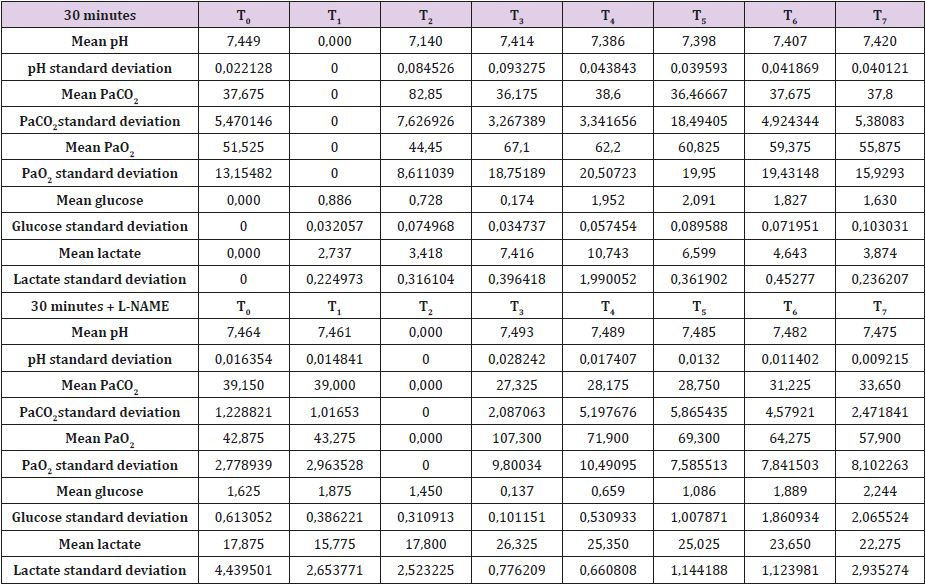
Table 5: SSS mean values and standard deviation for 30 minutes brain tamponade with and without L-NAME
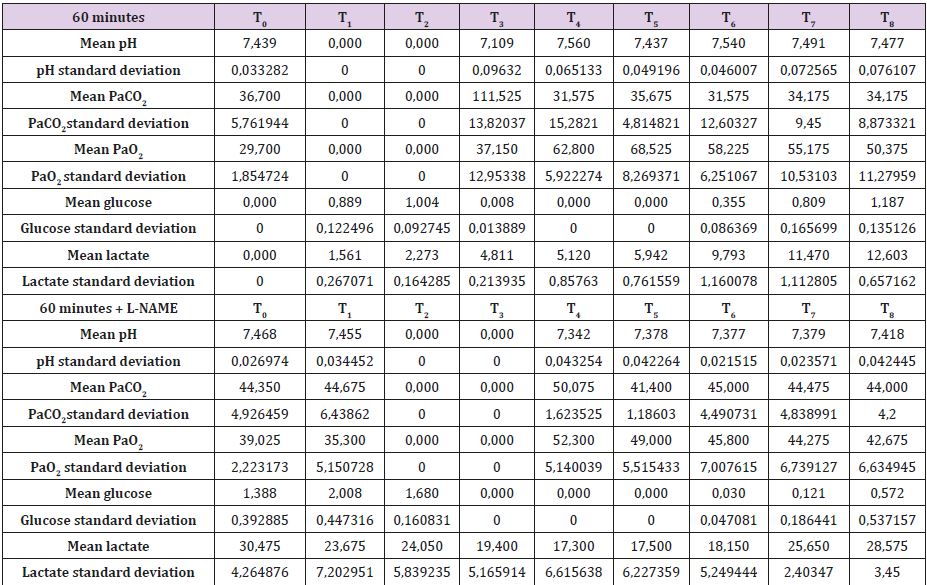
Table 6: SSS mean values and standard deviation for 60 minutes brain tamponade with and without L-NAME.
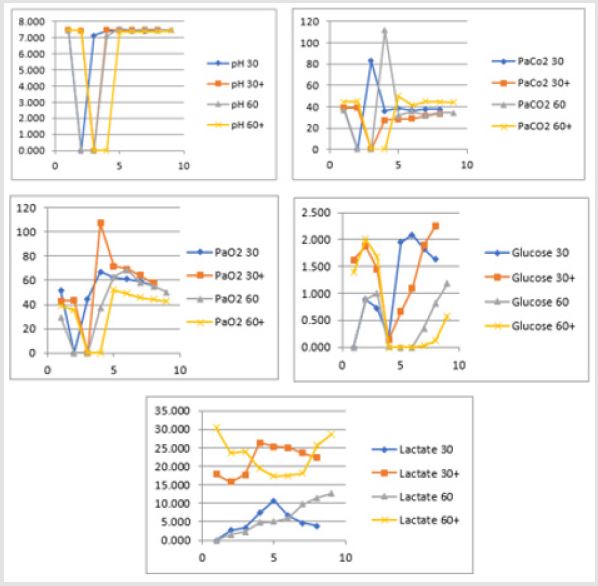
Figure 1: Trend in pH, PaCO2, PaO2, glucose and lactate during 30 and 60 minutes brain tamponade with and without L-NAME pretreatment.
Discussion
Brain metabolism is a complex mechanism in which glycolysis
and oxidative phosphorylation are the main protagonist in ATP
generation and thus energetic supply. During prolonged ischemia
such mechanism are impaired and there is a shift towards different
anaerobic pathways that only partially can maintain ATP formation.
In lasting ischemia, a progressive cellular breakdown, due to ATP
dependent pump blockage, lead to brain death. Most of the paper
are centered on focal ischemia secondary to the simplicity to
evaluate the condition as well as to the frequency of the condition.
On the other hand, prolonged global ischemia is hard to evaluate,
and it is hard to find clinical conditions in which proper evaluation
are feasible. For this very reason, in this paper we have used animal
specimens in which brain tamponade was achieved through ICP
increase. Such a choice was made not hinder our data with changes
in metabolism secondary to the tamponade etiology. Furthermore,
as our work intend to evaluate metabolite no clinical or neurological
evaluation has been carried on. Our experiment analyzed different
metabolic processes occurring during different length of brain
tamponade, namely 30 and 60 minutes of null CBF.
Our results reflect the shift from aerobic metabolism to
anaerobic one with consequent production of acidic metabolite.
Such a modification is visible from the progressive increase of
the PaCO2 SSS in contraposition with the stability of the arterial
PaCO2 Art that tend to remain stable. It is the manifestation of the
progressive glycolytic metabolism and consumption of the stored
glucose. In the same way, PaO2 SSS decrease while PaO2 Art tend
to be 15% higher than baseline in the attempt to correct the lack
of brain oxygen. Being impossible during tamponade to measure
venous contempt and thus brain metabolite, microdialysis seems
to be the only way to monitor what happen during tamponade. The
drop-in glucose levels are the hallmark of glycolysis; such a drop
is made even bigger from the impossibility for “fresh” glucose to
arrive through the blood stream. Nonetheless, before a complete
glucose depletion all the glucose supply is consumed. Such supplies
are stored in the intracellular space. With glucose reduction comes
pyruvate and lactate accumulation. In the meanwhile, the stability
in glycerol levels suggest that no cell membrane damage occur
during this phase.
Analyzing those data, it can be inferred that the brain, after 30
minutes of tamponade, is not metabolically dead as it continues
to burn substrate to produce energy for its maintenance. This
tendency is even more evident after reperfusion. Brain cells start
again to use blood-carried glucose and the pyruvate produced during the ischemic interval to support its metabolism leading to
even higher level of lactate. In this phase, ischemia related severe
mitochondrial damage lead to an increase in glycerol levels with
a resulting slowing down in aerobic metabolism recovery. As a
further sign of metabolic reversibility, EEG can be recorded again
unlike during tamponade. The 60 minutes tamponade is a totally
different situation. In fact, in this scenario intracellular glucose and
pyruvate are completely used up leading to an increase in lactate
levels that reach its highest after 90 minutes from reperfusion.
Moreover, lactate/pyruvate ratio starts to increase, and pH levels
rise due to the displacement towards acidotic metabolism despite
the changes in O2 and CO2 levels. Those data represent the decay of
all the back- up mechanism described in the 30 minutes tamponade
scenario. Furthermore, there is a shift in metabolism towards
NO production with an ulterior consequent decrease of oxidative
phosphorylation that leads to a worsening in cellular capability to
confront global ischemia.
Sign of the severity of the condition is the absence of electrical EEG recovery at the end of brain tamponade. This data seems to suggest that a longer global brain ischemia has an important effect on how cerebral cells try to preserve function during reperfusion. Nonetheless, even though brain cells cease their electrical activity they seem to be still metabolically “alive”. These data are of the utmost importance in defining irreversible cerebral damage and more in general brain death. Nowadays, the internationally accepted medical criteria for detecting brain death, both for the American “whole brain” [15] and the British “brain stem death” doctrine [16], are based on the finding of two isoelectric lines in two EEGs at least 24 hours apart. As previously suggested the electrical measurement only address part of the problem. Even though a metabolically active brain is not the same as a functioning brain we suggest that adding metabolite analysis to our brain monitoring can be useful in discriminating the window between brain ischemia and the start of electrical inactivity.
In our opinion, the use of microdialysis [17,18] can provide a more accurate evaluation during monitoring for intensive care unit patients in comparison with other techniques (ICP monitoring [19], brain oxygenation [17,18,20], transcranial Doppler ultrasound [17,18] or scalp EEG [21]). In fact, despite its minimally invasiveness, it gives an “instantaneous picture” of brain metabolism. An even more interesting data supporting the persistence of active brain metabolism during brain tamponade is given from the results of the L-NAME pretreatment experiment. NO and its metabolites are involved in several detrimental pathways, such as apoptotic signals, inflammatory mediators and activation of proteinase enzymes. Also, combination of NO and superoxide anion leads to the formation of a very toxic compound of piroxantrone, which yields to the protein nitro tyrosination and cell death. Finally, proximitized can distribute its poisonous effects into neighboring cells and tissues [14]. In addiction NO increase seems to disrupt the blood brain barrier creating even more damage. For this very reason, L-NAME was chosen to better understand if in ischemic brain NO reduction can affect brain metabolism during tamponade. It seems that after L-NAME pretreatment there is a switch to a “partial aerobic glycolysis” as previously reported by Regli [2,10].
This process seems testified by the increase in pyruvate that is used in the mitochondria to ensure energetic metabolism. Furthermore, the aforementioned interference with NO and ONOO- radical formation lead to the maintenance of creatinine phosphatase activity adding, as a support in cellular metabolism, the effect of the creatinine-phosphate/creatinine system (CrP/Cr). This system has the double role of ATP producer and mitochondrial support in dealing with ischemic damage, thus preserving oxidative phosphorylation [11,12]. Such findings seem to suggest that an organ capable of changing its metabolism can’t be “dead” as previously thought. In addition, EEG evaluation in L-NAME pretreated animals follows what seen in metabolism. In fact, while in basal condition after 60 minutes of brain tamponed there is no recover in electrical activity, in treated one’s partial recover can be witnessed. Though interesting such data are of difficult interpretation being taken from animal samples but remain an intriguing one. Such findings are obviously far from clinically useful but may open a discussion on whenever considering the brain dead. Furthermore, a suggestion in implementing ICU monitoring with microdialysis is warranted as a way to better monitor patients as well as a way to collect more human data to achieve a better comprehension of this complex problematic.
Conclusion
Metabolic changes during prolonged global brain ischemia are yet to be clearly defined. Despite cortical activity disappearance, brain metabolism seems to remain active especially in the first 30 minutes of ischemia. Such finding might change the way we define brain death and, in the future, might increase the possibility for neuroprotection. In this contest, brain microdialysis may represent a much more accurate monitoring system for neurological intensive care unit patients than the one of common use. Furthermore, pharmacological pretreatment might, with further studies, modify brain metabolism during tamponade increasing the gap between brain lesions and brain death. Obviously, there is still lot to investigate and many aspects of brain metabolism are still prone to be addressed by further studies, but our data might represent a starting point in a better definition of brain death and irreversible brain damage.
Declarations of Interest
The authors declare that they have no conflict of interest.
References
- Kristián T (2004) Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium 36(3-4): 221-233.
- Winn HR (2016) Youmans and Winn Neurological Surgery. In Winn HR (Eds.), (7th), New York, USA, 14.
- Hossmann KA, Grosse Ophoff B (1986) Recovery of monkey brain after prolonged ischemia. I. Electrophysiology and brain electrolytes. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 6(1): 15-21.
- Awooda HA, Lutfi MF, Sharara GM, Saeed AM (2013) Role of N-Nitro-L-Arginine-Methylester as antioxidant in transient cerebral ischemia and reperfusion in rats. Exp Transl Stroke Med 5: 1.
- Back T, Hemmen T, Schüler OG (2004) Lesion evolution in cerebral ischemia. J Neurol 251(4): 388-397.
- Hossmann KA, Fischer M, Bockhorst K, Hoehn Berlage M (1994) NMR imaging of the apparent diffusion coefficient (ADC) for the evaluation of metabolic suppression and recovery after prolonged cerebral ischemia. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 14(5): 723-731.
- Derbyshire DR, Clark RG (1980) Cerebral recovery after prolonged global brain ischaemia. Lancet Lond Engl 2: 637.
- Imai H, Konno K, Nakamura M, Shimizu T, Kubota C, et al. (2006) A new model of focal cerebral ischemia in the miniature pig. J Neurosurg 104(Suppl-2): 123-132.
- Bahlmann L, Klaus S, Baumeier W, Schmucker P, Raedler C, et al. (2003) Brain metabolism during cardiopulmonary resuscitation assessed with microdialysis. Resuscitation 59(2): 255-260.
- Regli L, Held MC, Anderson RE, Meyer FB (1996) Nitric oxide synthase inhibition by L-NAME prevents brain acidosis during focal cerebral ischemia in rabbits. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 16(5): 988-995.
- Li T, Wang N, Zhao M (2012) Neuroprotective effect of phosphocreatine on focal cerebral ischemia-reperfusion injury. J Biomed Biotechnol 2012: 168756.
- Tang LH, Xia ZY, Zhao B, Wei XD, Luo T (2011) Phosphocreatine preconditioning attenuates apoptosis in ischemia-reperfusion injury of rat brain. J Biomed Biotechnol 2011: 107091.
- Mohammadi MT, Dehghani GA (2015) Nitric oxide as a regulatory factor for aquaporin-1 and 4 gene expression following brain ischemia/reperfusion injury in rat. Pathol Res Pract 211(1): 43-49.
- Mohammadi MT (2016) Overproduction of nitric oxide intensifies brain infarction and cerebrovascular damage through reduction of claudin-5 and ZO-1 expression in striatum of ischemic brain. Pathol Res Pract 212(11): 959-964.
- Truog RD, Fletcher JC (1990) Brain death and the anencephalic newborn. Bioethics 4(3): 199-215.
- Sundin Huard D, Fahy K (2004) The problems with the validity of the diagnosis of brain death. Nurs Crit Care 9(2): 64-71.
- Bhatia A, Gupta AK (2007) Neuromonitoring in the intensive care unit. I. Intracranial pressure and cerebral blood flow monitoring. Intensive Care Med 33(7): 1263-1271.
- Bhatia A, Gupta AK (2007) Neuromonitoring in the intensive care unit. II. Cerebral oxygenation monitoring and microdialysis. Intensive Care Med 33(8): 1322-1328.
- Harris CH, Smith RS, Helmer SD, Gorecki JP, Rody RB (2002) Placement of intracranial pressure monitors by non-neurosurgeons. Am Surg 68(9): 787-790.
- Narotam PK, Morrison JF, Nathoo N (2009) Brain tissue oxygen monitoring in traumatic brain injury and major trauma: outcome analysis of a brain tissue oxygen-directed therapy. J Neurosurg 111(4): 672-682.
- Stuart RM, Schmidt M, Kurtz P, Waziri A, Helbok R, et al. (2010) Intracranial multimodal monitoring for acute brain injury: a single institution review of current practices. Neurocrit Care 12(2): 188-198.
