 Case Report
Case ReportAbstract
Objective: Type 1 Diabetes (T1DM) is an autoimmune disease. Children affected by T1DM are at higher risk of other autoimmune comorbidities and the combination of two or more organ specific disorders should define the diagnosis of Autoimmune Polyglandular Syndrome (APLS).
Case Report: Here we report the case of a young girl affected by an atypical APLS type 3. She had a diagnosis of T1DM at 4 years old, celiac disease at 6 years and autoimmune thyroiditis at 7 years. Her personal history was complicated by necrotizing glomerulonephritis ANCA-associated with important effects on glycaemic control and other autoimmune comorbidities.
Conclusion: we analyse a multifaceted case, in a very young girl, characterized by the coexistence of different autoimmune diseases influencing endocrine functions and metabolic control, during the different phases of a complex therapy.
Keywords: Type 1 Diabetes; Autoimmune Polyglandular Syndrome; Necrotizing Glomerulonephritis ANCA-Associated
Introduction
Type 1 Diabetes (T1DM) is an autoimmune, multifactorial, disease caused by beta cells destruction induced by immune response [1]. The pathophysiology of T1DM is still unclear but genetic predisposition is a landmark of this condition. HLA and non-HLA genes are involved in the risk of autoimmunity onset and many of these genes are shared by different autoimmune settings; thus, children affected by T1DM are at increased risk of other autoimmune comorbidities[2,3].In particular, T1DM could be part of autoimmune polyglandular syndrome cluster.
In 1980 Neufled and Blizzard proposed a classification of the autoimmune polyendocrine syndromes (APS) Table 1 [4]. These conditions are characterized by lack of self-tolerance and by induction of immune responses directed against self-structures with the coexistence, in the same patient, of more than one autoimmune disease [5]. The term polyendocrine is not completely appropriate, because several non-endocrine scenarios could be part of the clinical picture of these autoimmune syndromes [5,6]. Here, we report the case of a girl affected by APS type 3 and the course of the different diseases’ diagnosis in her young life.
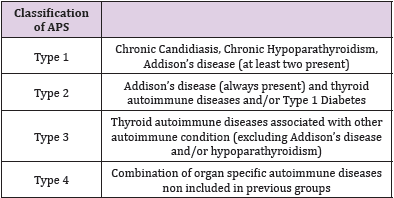
Table 1: Neufled and Blizzard’s classification of APS [4].
Case Report
MF, female, was born at 38 weeks by vaginal delivery. Pregnancy was complicated by placenta disruption at 18 weeks. Birth weight was appropriate for gestational age. Perinatal period was uncomplicated. Familial history was negative for particular medical condition and consanguinity. At 4 years of age, MF had diagnosis of T1DM complicated by ketoacidosis (pH 7.2, HCO3- 11.7 mmoL/L, HbA1c163 mmol/mol). Multi-daily injections therapy, according to basal bolus insulin regimen, was started with a good metabolic control. At 6 years of age, MF complained diarrhoea and abdominal pain. Celiac screening was positive; endoscopy confirmed the diagnosis of celiac disease with type 3 histology according Marsh- Oberhuber classification. Gluten Free Diet (GFD) was then switched on with a good compliance.A transient phase of hyperthyroidism was recorded when MF was 7 years old: TSH was 0.007 mcUI/ mL, FT4 2.3 ng/dL, anti-thyroglobulin antibodies (ATG) were positive 211.4 UI/mL (normal value < 29), thyrotropin receptor antibodies (TrAbs) and anti-thyroid peroxidase antibodies (ATP) were negative. Ultrasonography showed signs of thyroiditis. A new autoimmune diagnosis was made autoimmune thyroiditis.
She didn’t need treatment until 8 years of age, when MF presented clinical signs of hypothyroidism confirmed by laboratory findings (TSH 21.93 mcUI/mL, FT4 0.65 ng/dL) and so levothyroxine was started with at an initial dose of 0.5 mcg/kg/die. She had a second important symptomatic phase of hyperthyroidism at age of 10 years, under thyroxine therapy, and beta blocker drug (propranolol) was used. Blood examinations confirmed TrAbs negativity with TSH 0.02 mcUI/mL and FT4 3.09 ng/dL. Thyroxine treatment was stopped for few weeks. After few months, MF complained arthralgia and swelling of the hands with clinical signs of arthritis. Laboratory evaluation showed a slight increase of Anti-Nuclear Antibodies (ANA 1:80, homogeneous pattern) with negativity of inflammation indexes. Articular ultrasound was performed with normal imaging. HLA typing was HLA DRB1 *04 *16, HLA-A *03 *30, HLA-B *14 *49, HLA-C *07 *08, DQB1 *03 *05, HLA-DQA1 *01:02, HLA-DQA1 *03:03.
Treatment with Nonsteroidal anti-inflammatory drug was started for 3 weeks with good clinical response, but the symptomatology relapsed, after the withdrawal. A new complete evaluation was performed at Paediatric Rheumatologic Hospital with diagnosis of “Transient arthralgia and subcutaneous edema” in spite of an increased level of ANA titre (ANA 1:320, homogeneous pattern). During the follow up period, MF showed also persistent microalbuminuria although good glycaemic control. Proteinuria in 24 hours was normal in repeated controls, until the appearance of pathologic levels (800 mg/24h) deserving further investigations. The new findings were positivity for anti-neutrophil cytoplasmic antibodies (ANCA): subtype anti Myeloperoxidase (MPO), antiproteinase 3 negative. In May 2016 renal biopsy was performed and histology defined the diagnosis of necrotizing glomerulonephritis ANCA-associated. Optic microscopy showed focal and segmental mesangial hypercellularity, one small area of fibrinoid necrosis and one cellular crescent. Tubules and interstitium were normal. Immunofluorescence showed IgG, IgM and C3 mesangial and parietal traces. The diagnosis was renal-limited vasculitis; MF was 11 years old.
Treatment was started with methylprednisolone in bolus for three days at dosage of 10 mg/kg/die, followed by oral prednisone at 1 mg/kg/die for one month and then tapered in the following weeks. She had no good response with this first line therapy with persistent high proteinuria levels (703.57 mg/24 h). Second line therapy was started with cyclophosphamide at the dose of 2 mg/kg/die for three months, during corticosteroid tapering, with progressive reduction of proteinuria (402.5 mg/24 h, 242.1 mg/24 h). Then mycophenolate mofetil was started as long term consolidation therapy at a mycophenolate as “600 mg/mq/die administered twice daily, with maintenance of negative proteinuria, although the dose was subsequently reduced for the onset of leukopenia [7,8]. During the follow up period, the girl presented some other transient phases of hyper- and hypo-thyroidism, in spite of the compliance to GFD. The strange course of thyroid autoimmunity during these phases is described in Figure 1.
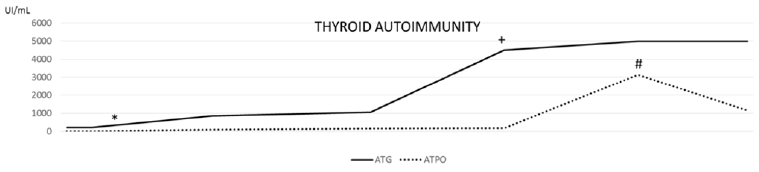
Figure 1: Thyroid autoimmunity course during the follow up (ATG anti thyroglobulin antibodies, ATPO anti thyroid peroxidase antibodies, x axis: timing): * Autoimmune Thyroiditis diagnosis, + arthralgia, # Glomerulonephrytis diagnosis.
Discussion
Type 1 Diabetes is an autoimmune disease in which genetic and environmental factors interact to define not only determinism of its pathophysiology but also the susceptibility to other autoimmune conditions [3].
Often the cluster of autoimmune diseases could be present in the same person and so the diagnosis of autoimmune polyglandular syndrome could be made [9]. In our patient, Autoimmune Polyglandular Syndrome type 3 was diagnosed, but there are some concerns about. Neufled and Blizzard classification of APS was completed by Betterle in 2002 (Table 2); in particular, according to the latter, our clinical case could not be placed in a defined condition: MF had autoimmune diabetes, autoimmune thyroiditis, celiac disease and vasculitis, so she could be diagnosed as APS type 3a, 3b or 3d [4,10]. Eisenbarth et al in 2004 underlined the usefulness of a simpler classification for these autoimmune conditions based on genetic background, age at onset and clinical settings to avoid mistakes and doubts [5].However, beyond all type of classification, our patient’s history was characterized by an uncommon association in her clinical picture.
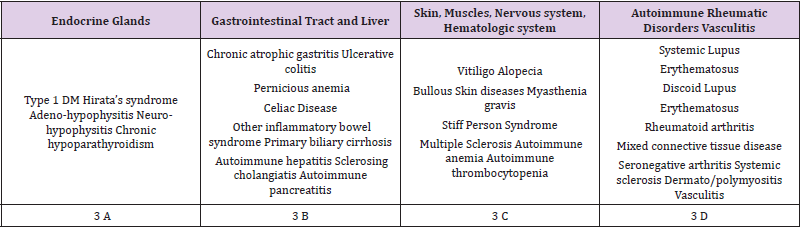
Table 2: Type 3 APS according to Betterle classification: the diagnosis requires the presence of autoimmune thyroid disease with other autoimmune conditions [9].
ANCA have been detected more frequently in patients with a history of certain autoimmune diseases. Such an association may simply reflect an underlying tendency to immune system dysfunction in these patients and the finding of positive ANCA serology does not reliably herald the development of ANCAassociated vasculitis. In our patient ANCA was searched in the autoimmunity panel after the detection of pathologic proteinuria in urine, despite of a good glycaemic control, and correlate with histologic findings at renal biopsy. Few cases of ANCA vasculitis in autoimmune poly endocrinopathy syndrome have been described and this is the first case of renal vasculitis in a patient affected by Autoimmune Polyglandular Syndrome type 3, to our knowledge. Recent evidences underline the importance of HLA genotype in the susceptibility to MPA: gene variants appear to alter immune cell proteins and responses, becoming key factors in the pathogenesis of ANCA-associated vasculitis [11]. In particular, the patient HLA genotype could explain her susceptibility to ANCA associated vasculitis [7]. MF is now adolescent and her clinical history is, probably, similar to an iceberg in which we can observe only the visible part, we don’t know the submerged one at this time and we don’t know what else could happen in her next future.
In particular, one important teaching from this case is the influence and complex interactions of autoimmunity on endocrine functionality. At first, the strange instability of thyroid function was one prominent feature in our patient. Transient phases of hypothyroidism and hyperthyroidism were recorded: each phase was accompanied by modification in thyroid autoimmunity subset, as showed in Figure 1, and every phase of hypothyroidism was preceded by hyperthyroidism, as to indicate an autoimmune attack to the gland. Probably, the autoimmune system, the therapies used, and the stress could be the reading key of these complex immuneendocrine interactions. These might lead to the lack of recovery equilibrium typical of autoimmune thyroiditis course [12]. The effects of autoimmune comorbidities on glycaemic control has also been experimented: hypoglycemia and hyperglycemia were clearly complications observed in our patient management [13]. Last but not least, moreover, this lesson: Nephropathy in Diabetic Children is not only «Diabetic». Renal biopsy played a fundamental role in the diagnosis, prognosis and in our therapeutic decisions [14]. Our case report highlights the importance of a strict clinical and laboratory monitoring in these complex pediatric patients, in a multidisciplinary approach, giving the right attention to the appearance of new signs or symptoms, even if they could be related to the first diagnosis.
Funding Source
No external funding for this manuscript.
Financial Disclosure
The authors have no financial relationships relevant to this article to disclose.
Conflict of Interests
The authors have no potential conflicts of interest to disclose.
References
- Lenmark A, Larsson HE (2013) Immune therapy in type 1 Diabetes Mellitus. Nature Reviews Endocrinology 9(2): 92-103.
- Sharma A, Liu X, Hadley D, Hagopian W, Chen WM, et al. (2018) Identification of non-HLA genes associated with development of islet autoimmunity and type 1 diabetes in the prospective TEDDY cohort. J Autoimmun : pii: S0896-8411(17): 30739-30744.
- Kakleas K, Soldatou A, Karachaliou F, Karavanaki K (2015) Associated autoimmune diseases in children and adolescents with type 1 diabetes mellitus. Autoimmun Rev 14(9): 781-797.
- Neufeld M, Blizzard RM (1980) Polyglandular autoimmune diseases. In: Pinchera A, Doniach D, Fenzi GF, Baschieri L (Eds.) Symposium on autoimmune aspects of endocrine disorders. Academic Press 1980, New York, USA, pp. 357-365
- George S Eisenbarth, Peter A Gottlieb (2004) Autoimmune Polyendocrine Syndromes. N Engl J Med 350: 2068-2079.
- Betterle C, Garelli S, Coco G, Burra P (2014) A rare combination of type 3 autoimmune polyendocrine syndrome (APS-3) or multiple autoimmune syndrome (MAS-3). Auto Immun Highlights 5(1): 27-31.
- Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, et al. (2012) Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med 367(3): 214-223.
- Plumb la, Oni L, Marks SD, Tullus K (2018) Paediatric anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis: an update on renal management. Pediatr Nephrol 33(1): 25-39.
- Hansen MP, Matheis N, Kahaly GJ (2015) Type 1 diabetes and polyglandular autoimmune syndrome: A review. World J Diabetes 6(1): 67-79.
- Betterle C, Dal Pra C, Mantero F, Zanchetta R (2002) Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev 23(3): 327-364.
- Merkel PA, Xie G, Monach PA, Ji X, Ciavatta DJ, et al. (2017) Vasculitis Clinical Research Consortium. Identification of Functional and Expression Polymorphisms Associated with Risk for Antineutrophil Cytoplasmic Autoantibody-Associated Vasculitis. Arthritis Rheumatol 69(5): 1054-1066.
- Radetti G, Maselli M, Buzi F, Corrias A, Mussa A, et al. (2012) The natural history of the normal/mild elevated TSH serum levels in children and adolescents with Hashimoto’s thyroiditis and isolated hyperthyrotropinaemia: a 3-year follow-up. Clin Endocrinol (Oxf) 76(3): 394-398.
- Krzewska A, Ben-Skowronek I (2016) Effect of Associated Autoimmune Diseases on Type 1 Diabetes Mellitus Incidence and Metabolic Control in Children and Adolescents. Send Biomed Res Int: 6219730.
- Radhakrishnan J, Cattran DC (2012) The KDIGO practice guideline on glomerulonephritis: reading between the (guide)lines--application to the individual patient. Kidney Int 82(8): 840-856.
