 Case Report
Case ReportAbstract
Introduction: Malformations in cortical development are due to anomalies during the formation of the cortical plaque in intrauterine life. These changes can occur in the stages of proliferation, migration or organization. Abnormal neuronal migration results in heterotopy. Periventricular nodular heterotopy consists of the atypical presence of gray matter in the periventricular white matter. It is estimated that approximately 80% of patients with this malformation develop epilepsy. Bearing in mind that epileptic seizures tend to significantly affect the quality of life of patients, understanding the severity and consequences associated with cortical malformations becomes essential to direct medical care. Thus, the objective of the present article is to report the case of a patient with epilepsy associated with periventricular nodular heterotopy, and through this to propose a discussion regarding the main evidence available in the literature that touches both clinical conditions.
Case Report: LML, a 31-year-old woman with a history of tonic-clonic seizures that began 2 years ago. Magnetic resonance imaging of the skull showed nodules with a signal similar to the gray matter diffusely distributed below the eppendymum, suggesting heterotopy.
Discussion: Although heterotopic nodules seem to act as a focus of abnormal neuronal activity, their role in epileptogenicity remains controversial. While electrophysiological studies demonstrate that they can be intrinsically epileptogenic, others indicate that a large epileptic network, rather than just a nodule, is responsible for epileptogenicity.
Conclusion: Malformations in cortical development represent an important comorbidity associated with the genesis of epilepsy. Even so, prospective electrophysiological studies are necessary to elucidate the pathophysiological mechanisms involved in this process.
Introduction
Cortical development malformations (CDM) consist of anomalies that arise from the interruption of the normal stages of formation of the cortical plaque [1]. In embryonic development, the human cortex develops through overlapping stages during the first two gestational trimesters [1]. These steps start with the proliferation and differentiation of neurons, which migrate until they finally organize themselves in the cortex [1]. Any abnormality in these stages can trigger changes in neuronal circuits, predisposing to several clinical manifestations, with epileptic seizures being the most common manifestations [1]. Epileptic seizures can produce variations in consciousness, in addition to sensory and motor variations [2]. Among the main cortical malformations associated with these seizures, heterotopias stand out. Heterotopies consist of abnormalities in cell migration, in which mature neurons do not complete their displacement to the cortex, culminating in the erroneous location of gray matter [3-5]. These MDC are named according to their location, which can be adjacent to the lateral ventricles, in the subcortical or leptomeningeal region [3-5].
Periventricular nodular heterotopia (PNH) can be unilateral or bilateral, isolated or multiple and located anywhere along the lateral ventricles [6]. Macroscopically, this condition corresponds to nodular-looking masses formed by adjacent gray matter or protruding into the lateral ventricles [6]. These nodules can be either single or multiple, both separate and contiguous [6]. It is estimated that about 80 to 90% of patients with periventricular nodular heterotopy develop epilepsy, the majority with multiple types of partial seizures [6,7]. Considering that epileptic seizures can significantly affect the quality of life of patients, understanding the severity and consequences associated with cortical malformations is essential to direct medical care. Thus, the objective of this article is to report the case of a patient with epilepsy associated with periventricular nodular heterotopy, and through this to propose a provocative discussion as to the main evidence available in the literature that touches both clinical conditions.
Case Report
LML, a 31-year-old woman, attended the medical consultation reporting episodes of tonic-clonic seizures that began 2 years ago. She denies having had previous treatment for seizures. There was a diagnosis of migraine since childhood. The physical examination showed no changes. The EEG was normal, but the magnetic resonance of the skull showed nodules of a signal similar to the gray substance diffusely distributed below the eppendymum, determining lobulations in the contours of the lateral ventricles, suggesting heterotopia (Figure 1). Treatment with lamotrigine and folic acid was started, with no new convulsive episodes since then.
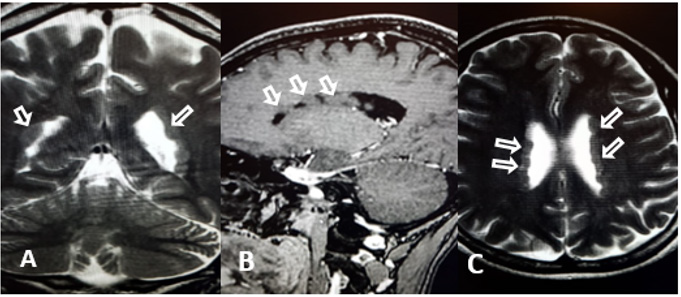
Figure 1: MRI - nodules of a gray matter-like signal diffusely distributed below the eppendyme, suggesting heterotopy. A - CORONAL; B - SAGITAL, C – AXIAL.
Discussion
coordinated manner, through a series of morphological transformations during embryonic development, going through well-defined stages of cell proliferation, migration and organization [3-5]. Cellular migration is dynamic and starts from the fifth week of gestation, when the telencephalic vesicle appears [3- 5]. Subsequently, the proliferation and initial differentiation of neuronal and glial precursors occurs [3-5]. Then, they move from the periventricular region to the cortical plate [3-5]. These phenomena are guided by the irradiated glial cells, which act as a support and guide for the correct location of neurons [3-5]. Abnormal neuronal migration results in heterotopia. The nodular heterotopy of the gray matter (HN) is a not uncommon condition in the brain and is most often located in the periventricular or subcortical white matter [6]. The microscopic aspect of HN comprises clusters of irregular nodules that separate through myelinated fibers, and can involve both neurons and glial cells, with a variable pattern of organization [1,6]. This condition can occur in isolation, concomitantly with other anomalies of cortical development or even associated with multiple congenital anomalies syndrome [6].
Periventricular nodular heterotopia (PNH) can be located anywhere along the lateral ventricles and can be unilateral or bilateral [8]. Typical bilateral PNH has already been reported in association with hypoplasia of the corpus callosum and cerebellum [1,9,10]. Although PNH is often limited to the periventricular region, it can occasionally lead to the formation of a larger mass, and consequently, trigger deformities or displacement of the lateral ventricle [1]. Mutations of the filamin A gene (FLNA), which is located on the long arm of the X chromosome, are considered the classic cause of PNH, although this condition can also occur sporadically or associated with other genetic mutations [11,12]. The FLNA gene plays a fundamental role for efficient cellular motility, given that it binds to actin, promoting networks at the front edge of mobile cells, and also binds to support cells until the signal necessary for cell locomotion [13,14]. Mutations in this protein can alter its functional capacity, triggering the atypical neuronal migration in bilateral PNH [13]. Epidemiologically, PNH has a higher prevalence in females, which can be associated with the fact that mutations in FLNA, the most common cause of injury, are considered lethal in males [13,15].
Periventricular nodular heterotopia is the main cortical malformation associated with convulsive disorders [3,8,9]. It is estimated that about 80 to 90% of patients with PNH develop epilepsy [6,7]. Epilepsy is characterized by periodic and paroxysmal changes in cortical and subcortical functions, which temporarily interrupt normal brain function [2]. When associated with PNH, this condition usually starts in the first years of life and can vary in severity. A study by Pisano et al., with 50 PNH patients, showed a prevalence of treatment-resistant epilepsy in about 56% of patients [10]. In the same study, the authors suggest that these patients may present any type of seizure, although complicated and treatment-resistant partial seizures represent the most common form in individuals with PNH [10]. As for the association between PNH and epilepsy, it is believed that heterotopic nodules act as a focus of abnormal neuronal activity. Even so, the role of these nodules in the genesis of epilepsy remains controversial. While electrophysiological studies demonstrate that they can be intrinsically epileptogenic, others indicate that a large epileptic network, instead of just a nodule, is responsible for epileptogenicity [16].
A study by Hui Ming Khoo et al. evaluated the functional internodular connectivity in heterotopy-related epilepsy and suggested that the cortical heterotopic nodules are functionally connected through synapses, or that they connect to a third common area, without one node being directly connected to the other [16]. These connections would represent the ability of these nodules to initiate epileptic seizures or to spread rapidly through them [16,17]. The authors suggest that ictal activity spreads more quickly between connected nodes than between unconnected nodes [16]. Through these findings, the authors believe that internodular connections are responsible for providing a path that facilitates the propagation of ictal activity [16].
Conclusion
Malformations in cortical development represent an important comorbidity associated with the genesis of epilepsy. Individuals with periventricular nodular heterotopy can present any type of seizure, with complicated and treatment-resistant focal crises being the most common. Although it is believed that the functional connections between heterotopic nodules are related to epileptogenicity, or greater ictal spread, prospective electrophysiological studies are still needed to elucidate this scenario.
References
- Leventer RJ, Guerrini R, Dobyns WB (2008) Malformations of cortical development and epilepsy. Dialogues Clin Neurosci 10(1): 47-62.
- PORTO Lívia Amorim, Jullyana de Souza Siqueira, Luciene Nascimento Seixas, Jackson Roberto Guedes da Silva Almeida,Lucindo José Quintans-Júnior, et al. (2007) The role of ion channels in epilepsy and considerations about antiepileptic drugs - a brief review. Journal of Epilepsy and Clinical Neurophysiology 13(4): 169-175.
- Lerman-Sagie T, Leibovitz Z (2016) Malformations of Cortical Development: From Postnatal to Fetal Imaging. Canadian Journal of Neurological Sciences / Journal Canadien des Sciences Neurologiques 43(5): 611-618.
- Murilo S MenesesI, Amanda Hertz, Christiane Gruetzmacher, Samanta F Blattes, Erasmo Barros da Silva Júnior, Ronaldo A Vosgerau, et al. (2006) Epilepsy and malformations of cortical development disorders. Journal of Epilepsy and Clinical Neurophysiology 12(3): 149-154.
- LEITE, Joana Sousa (2009) Refractory epilepsy due to malformations of cortical development: a descriptive study of 4 cases.
- Harding BN (1996) Gray matter heterotopy. In: Guerrini R, Andermann F, Canapicchi R, et al. (Eds.), Dysplasias of Cerebral Cortex and Epilepsy.1ª (edn.). Filadélfia, Pa: Lippincott-Raven 1996: 81-88.
- Dubeau F, Tampieri D, Lee N, Andermann E, Carpenter S, et al. (1995) Periventricular and subcortical nodular heterotopia. A study of 33 patients. Brain 118(5): 1273-1287.
- Ihle-Hansen H, Lohne SMH, Dahl-Hansen E (2019) Periventrikulær nodulær heterotopi [Periventricular nodular heterotopia]. Tidsskr Nor Laegeforen 139(16).
- Ebetiuc I, Bulk S, Leroy P (2019) L’hétérotopie nodulaire pé Un cas pédiatrique [Periventricular nodular heterotopia: a pediatric case]. Rev Med Liege 74(7-8): 388-390.
- Pisano T, Barkovich AJ, Leventer RJ, Squier W, Scheffer IE, et al. (2012) Peritrigonal and temporo-occipital heterotopia with corpus callosum and cerebellar dysgenesis. Neurology 79(12): 1244-1251.
- Kothare SV, VanLandingham K, Armon C, Luther JS, Friedman A, et al. (1998) Seizure onset from periventricular nodular heterotopias: depth-electrode study. Neurology 51(6): 1723-1727.
- Hannan AJ, Servotte S, Katsnelson A, Sisodiya S, Blakemore C, et al. (1999) Characterization of nodular neuronal heterotopia in children. Brain 122 (Pt 2): 219-238.
- Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, et al. (1992) Actin-binding protein requirement for cortical stability and efficient locomotion. Science 255(5042): 325-327.
- Fox JW, Lamperti ED, Ekşioğlu YZ, Hong SE, Feng Y, et al. (1998) Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21(6): 1315-1325.
- Parrini E, Conti V, Dobyns WB, Guerrini R (2016) Genetic Basis of Brain Malformations. Mol Syndromol 7(4): 220-233.
- Khoo HM, von Ellenrieder N, Zazubovits N, Hall JA, Dubeau F, et al. (2019) Internodular functional connectivity in heterotopia-related epilepsy. Ann Clin Transl Neurol 6(6): 1010-1023.
- Cossu M, Fuschillo D, Cardinale F, Castana L, Francione S, et al. (2014) Stereo-EEG-guided radiofrequency thermocoagulations of epileptogenic grey-matter nodular heterotopy. J Neurol Neurosurg Psychiatry 85(6): 611-617.
