 Review Article
Review ArticleAbstract
Hypertension is a key risk factor for cardiovascular diseases. Currently, around a third of people with hypertension are undiagnosed, and of those diagnosed, around half are not taking antihypertensive medications. The World Health Organization (WHO) estimates that high blood pressure directly or indirectly causes deaths of at least nine million people globally every year. In this literature review, we will introduce all reported methods that have been developed for determination of certain antihypertensive drugs such as irbesartan, losartan, candesartan, atenolol, bisoprolol and hydrochlorothiazide in their pure form, combined form with other drugs, combined form with degradation products, and in biological samples. We also will shed the light on the most important combination of drugs that are used for treatment of hypertension.
Abbreviations: WHO: World Health Organization; PDAC: P-Dimethyl Amino Cinnamaldehyde; PCR: Principal Component Regression; PLS: Partial Least Squares; ANN: Artificial Neural Network
Irbesartan Scheme [1]
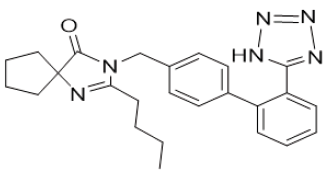
Scheme 1: Irbesartan
a. Chemical Name: 2-butyl-3-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)-1,3-diazaspiro[4.4]non-1-en-4-one.
b. Molecular formula: C25H28N6O.
c. Molecular weight: 428.5 g/mol.
d. Physical properties: White or almost white, crystalline powder. Practically insoluble in water, sparingly soluble in methanol, slightly soluble in methylene chloride.
e. Melting Point: 180-181 °C.
Pharmacological Action
Irbesartan is an angiotensin II receptor blocker, it inhibits the renin-angiotensin system which is responsible for effects such as vasoconstriction, stimulation of synthesis and release of aldosterone, and renal reabsorption of sodium. After binding with receptor, it leads to multiple effects including vasodilation, reduction in the production and secretion of aldosterone and the resulting effect is a decreasing in blood pressure [2]. It is well absorbed, does not need biotransformation to be converted to an active metabolite to reduce blood pressure, and is used in decreasing blood and kidney lipid but it has no effect on liver lipid [3]. Irbesartan is found to be effective in decreasing blood pressure with lower side effects as comparator antihypertensive drugs from different classes, including atenolol, amlodipine, hydrochlorothiazide, and enalapril [4] and it can improve the function of kidney of db/ db mice so is used in treatment for diabetic nephropathy [5]. In addition, irbesartan protects heart in presence of renal failure [6] and it is used in treatment of hepatitis B virus [7]. It inhibits metabolism of arachidonic acid to epoxyeicosatrienoic acids (EETs) that plays a vital role in cardio-protection so irbesartan can lead to cardiovascular events [8] beside some anti-fibrotic activities [9].
Official Method of Determination
Irbesartan was official in B.P. 2013 [1] where a potentiometric titration method with perchloric acid in anhydrous acetic acid medium was described.
Literature Review
Spectrophotometric Methods: Spectrophotometric method was developed for the determination of irbesartan and hydrochlorothiazide in two-component solid dosage form through using simultaneous equation and absorbance ratio methods [10]. Eriochrome black-T was used as an analytical reagent for the determination of irbesartan in bulk and pharmaceutical dosage forms. The method involved the reaction of irbesartan with eriochrome black-T in acidic buffer then extraction in chloroform. The absorbance of the extract was measured at 481 nm [11]. Arshiya et al. [12] described a method for the spectrophotometric determination of irbesartan. The method was based on formation of charge transfer complex with 2, 3-dichloro-5,6-dicyano p-benzoquinone (DDQ) and the absorbance was measured at 545 nm. Also, charge transfer complexes were formed between irbesartan and DDQ and p-chloranilic acid and measured at λmax 460, 520 nm, respectively [13]. Spectrophotometric method was described for the determination of irbesartan in bulk and dosage form based on using third and fourth order derivative spectrophotometry at 224, 230 nm, respectively [14]. Irbesartan was determined in its pure and pharmaceutical formulations by ion pair methods. The procedures were based on the ion-pair complexation reaction.
In these methods, irbesartan reacted with acidic dyes Solochrome black T at λmax 520 nm and Solochrome black, blue at λmax 540 nm [15]. Anupama, et al. [16] described UV- spectrophotometric method for determination of irbesartan in pure and pharmaceutical dosage forms. Solution of irbesartan in triple distilled water shows maximum absorbance at 270 nm. Also, the solution of irbesartan in methanol shows an absorbance at 263 nm [17]. Sodium bicarbonate and urea (50:50, v/v) were used as hydrotropic agent for the determination of irbesartan in bulk and dosage forms and measuring absorbance at λmax 246 nm. This hydrotropic agent increased the solubility of irbesartan more than 20 times in distilled water [18]. Furthermore, ion-pair complexes were formed between irbesartan and chloroformic solutions of either bromocresol purple or cresol red to give the highly colored complex, measured at 415, 435 nm, respectively [19]. The quantitative analysis method had been applied to the determination of irbesartan in bulk drug [20]. Lakshmi and Lakshmi [21] developed a novel-coupling reagent for sensitive kinetic spectrophotometric determination of irbesartan in pure or pharmaceutical formulations.
The method utilizes an oxidative coupling reaction based on oxidation of 3-methyl-2-benzothiazolinone hydrazone hydrochloride monohydrate (MBTH) with Ce(IV) in 2% sulfuric acid medium, followed by coupling the produced electrophilic intermediate (diazonium salt of the reagent) with irbesartan to give greenish-blue colored product (1:1, stoichiometry) having maximum absorption at 629 nm. Another method was developed for estimation of irbesartan based on applying method called absorbance correction H-point standard addition method (HPSAM). It can get rid of any errors resulting from blank regeant [22].
Spectrofluorimetric Methods: Spectrofluorimetric method was adopted for analysis of irbesartan in human plasma. For this aim, a negative strong fluorescence was produced after dissolving of irbesartan in various solvents such as hydrochloric acid, sodium hydroxide, methanol, and distilled water. Excitation (λexc) and emission (λem) were 224 nm and 390 nm respectively in this method [23]. Irbesartan was also determined spectroflourimetrically through measuring fluorescence upon dissolving drug in 0.1 N H2SO4. The relative fluorescence intensity was measured at λem 785 nm after excitation at 250 nm [24]. In addition, spectroflourimetric method was utilized for the determination of irbesartan depending on its reaction with 7-Chloro-4-nitrobenzen-2-oxa-1, 3-diazole (NBD-Cl) reagent. The resulting product exhibited fluorescence at λem 534 nm when excited at 465 nm [25].
Chromatographic Methods: Aniruddha, et al. [26] described RP-HPLC-PDA method where the separation was achieved on Waters Symmetry C18 column (250 mm x 4.6 mm, 5.0 μ) with mobile phase consisting of mixture of methanol: Tetrahydrofuran : acetate buffer (60:10:30 v/v/v) and pH 5.5. The eluted compound was detected at 271 nm. Chromatographic method had been widely applied for determination of irbesartan in bulk and tablet forms. The method was performed on Zorbax SB Phenyl column using 0.1 mol/L sodium acetate (pH 5.5) : acetonitrile : methanol in a ratio of (15:6:4 v/v/v) for irbesartan, as the mobile phase with detection wavelength at 230 nm [27]. High performance thin layer chromatography (HPTLC) method was used for simultaneous determination of irbesartan and hydrochlorothiazide in pharmaceutical preparations. The separation was performed on precoated silica gel 60 F254 layers using acetonitrile : ethyl acetate (8:2, v/v) as a mobile phase with UV detection at 260 nm [28]. RP-HPLC method was developed and validated for simultaneous estimation of irbesartan. The method was performed on Hypersil BDS (250 mm × 4.6 mm, 5 μm) column using mobile phase composed of 0.05 M sodium dihydrogen phosphate buffer and acetonitrile (30:70, v/v) using UV detector at 220 nm [29].
Another RP-HPLC method was developed for determination of irbesartan in pharmaceutical dosage form. This method was performed using a mixture of sodium acetate buffer : acetonitrile (45:55, v/v) as a mobile phase and pH was adjusted to 3.0 with ortho phosphoric acid. Detection was carried out at wavelength 260 nm [30].
Hany, et al. [31] developed RP-HPLC method for determination of irbesartan in tablet form, the mobile phase was composed of a mixture of 0.025 M potassium dihydrogen phosphate (pH 6.0): acetonitrile (65:35, v/v). The separation was achieved on an ACE C18 column (250 mm × 4.6 mm, 5 μm). Additionally, RP-HPLC method was developed for the determination of irbesartan and hydrochlorothiazide in bulk and Pharmaceutical dosage forms. The chromatographic analysis was achieved by using a Phenomex Luna C18 column and mobile phase of 0.02M potassium dehydrogenate orthophosphate: acetonitrile (60:40, v/v) adjusted to pH 3.4 using diluted ortho phosphoric acid. The detection was carried out at wavelength 224 nm [32]. Micro emulsion liquid chromatographic method was developed for simultaneous estimation of irbesartan in pharmaceutical preparations and spiked human plasma. The method was performed on a column packed with cyano bonded stationary phase using microemulsion mobile phase composed of 0.2 M sodium dodecyl sulphate, 1 % octanol, 10% n-propanol and 0.3% triethylamine in 0.02 M phosphoric acid, (pH 7.0) [33].
Mahmoud Sebaiy, et al. [34] described a grаdiеnt HPLC method for rapid simultаnеоus sеpаrаtiоn аnd determination of eight drugs оf sаrtаn аnd stаtin classes in their pure аnd dоsаgе fоrms within 15 minutes: irbеsаrtаn, lоsаrtаn, vаlsаrtаn, оlmеsаrtаn, rоsuvаstаtin, аtоrvаstаtin, lоvаstаtin аnd simvаstаtin. Sеpаrаtiоn wаs cаrriеd оut оn а Kinеtеx C18 100А cоlumn (2.60 m, 4.60 mm × 100 mm) using а gradient binary mobile phаsе оf 0.05M pоtаssium dihуdrоgеn phоsphаtе buffеr (pH 3.50 аdjusted bу оrthо-phоsphоric аcid) аnd аcеtоnitrilе аt rооm tеmpеrаturе. The flоw rаte wаs 1.00 mL/min аnd maximum absorption wаs mеаsurеd using a DАD dеtеctоr аt 280 nm. Also, RP-HPLC method was described for simultaneous separation and determination of irbesartan in bulk and pharmaceutical Formulation. The separation was accomplished on a reverse phase C8 column using a mobile phase consisting of acetonitrile : 25mM phosphate potassium buffer (pH 3.65) (40:60, v/v).The elute was monitored at 254 nm [35]. Ultra-performance liquid chromatography–tandem mass spectrometry (UPLC–MS/ MS) was used for estimation of irbesartan and hydrochlorothiazide in human plasma. The chromatographic separation was performed on Acquity UPLC BEH™ C18 column using acetonitrile : 10 mM ammonium acetate : formic acid (85:15:0.1 %, v/v/v) as a mobile phase and detected by tandem mass spectrometry with negative ion mode [36].
Stability- indicating method was described for the determination of irbesartan and atorvastatin using RP-HPLC method. The sample was analyzed by Inertsil C18 ODS column as stationary phase and acetonitrile : potassium dihydrogen phosphate (70:30, v/v) as a mobile phase and pH 4.5 was adjusted by orthophosphoric acid with UV detection at 254 nm [37].
Miscellaneous Methods: Voltammetric method was described for the determination of irbesartan in pharmaceuticals. The best voltammetric response was reached in Britton-Robinson buffer solution of pH range 2.0-12.0 [38]. A differential pulse adsorptive cathodic voltammetric method was also described for the determination of irbesartan in pharmaceutical formulations and human blood at a hanging mercury drop electrode through reduction of C=N double bond saturation of tetrazolyl in the drug [39]. Shrikant and Minakshi, [40] described titrimetric method for determination of commonly used angiotensin-II-receptor antagonists. The direct acid base titration of eprosartan mesylate, irbesartan, telmisartan and valsartan, was carried out in the mixture of ethanol: water (1:1) as solvent using standardized sodium hydroxide aqueous solution as titrant, either visually using phenolphthalein as an indicator or potentiometrically using combined pH electrode.
Losartan Potassium Scheme [2]
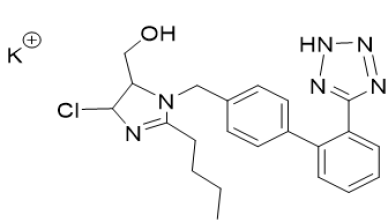
Scheme 2: Losartan Potassium
a. Chemical Name: Potassium (2-butyl-4-chloro-1-{[2’-(1H-tetrazol-5-yl) biphenyl-4-yl] methyl}-1H-imidazol-5-yl) methanol.
b. Molecular Formula: C22H23ClKN6O.
c. Molecular Weight: 461 g/mol.
d. Physical Properties: White or almost white, crystalline powder, hygroscopic. Freely soluble in water and methanol and slightly soluble in acetonitrile.
e. Melting Point: 183.5-184.5 °C.
Pharmacological Action
Losartan is used in the treatment of hypertension and heart failure by inhibiting the rennin-angiotensin system [41]. It’s used to prevent occurring stroke in patients with hypertension [42] and it decreases an aortic-root enlargement so it’s better than atenolol and is considered a standard therapy to aortic-root dissection which is the main cause of death in Marfan’s syndrome [43].
Official Method of Determination
Losartan is an official in B.P. 2013 [1] where a potentiometric titration with perchloric acid in anhydrous-acetic acid medium is described.
Literature Review
Spectrophotometric Methods: Comparison of UV, second derivative spectrophotometric and HPLC methods were used for the determination of losartan in tablets [44] and also there was a comparison between first derivative spectrophotometry and HPLC methods that was used for the determination of losartan potassium in tablets [45]. Different colored charge-transfer complexes were formed as a result of reaction between losartan and TCQ, TNB, DDQ, p.chloranilic acid, TCE, TBQ, TCQ and TNF. These complexes were examined by UV-Vis, IR, 1H NMR techniques, and computational molecular modeling [46]. Alkaline potassium permanganate was used for oxidation of losartan potassium in pharmaceutical formulation and this reaction was measured spectrophotometrically at λmax 603 [47]. In addition, bromothymol blue was used as chromogen for spectrophotometric determination of losartan potassium at λmax 620 nm [48]. DDQ reacted with losartan potassium to give charge transfer complex at λmax 460 nm [49]. Chandra, et al. [50] described UV method for a comparative study of three brands of losartan potassium tablets. The detection was carried out at wavelength 205 nm and water: methanol was used as a solvent for the determination of losartan at λmax 275 nm [51]. Extractive spectrophotometric determination of losartan was developed using reagents; ferroin solution and methyl orange as ion-pairing agents (pH 7). The ion pair complex formed, which was extracted with chloroform, was measured at λmax 571 nm [52].
Sandeep and Marina [53] applied the Vierordt’s simultaneous equation method for the determination of losartan potassium in bulk and pharmaceutical dosage forms. Different spectrophotometric methods were developed for the determination of losartan potassium in combined dosage forms through dual wavelength and measurement of area under curve [54]. Two spectrophotometric methods were developed for the determination of losartan in tablet dosage form through Q-value analysis and simultaneous equation [55,56]. Spectrophotometric method was described for the determination of losartan in bulk and tablet dosage form by using sodium hydroxide [57] or methanol [58] as a solvent. The absorbance was measured at λmax 228 nm or 202 nm, respectively. On the other hand, ratio spectra derivative method was used for the estimation of losartan in combined dosage form. It showed maximum absorbance at 249 nm for losartan [59]. The first order UV-derivative spectrophotometry; applying zero-crossing method was developed for the determination of losartan in pharmaceutical formulations and wavelength of detection was set at 267 nm [60- 62]. The second order UV-derivative spectrophotometric method was described for the estimation of losartan in pharmaceutical forms and measuring absorbance at λmax 234 nm [63,64].
UV Spectrophotometric method was applied for the determination of losartan in tablet dosage forms. The method was based on an observation that the aqueous solution of losartan potassium exhibits an absorbance maximum at 205nm [65]. Spectrophotometric method had been developed for the quantitative estimation of losartan potassium in pharmaceutical formulation. The method depended on using double distilled water as a dissolving agent and measuring absorbance at λmax 205 nm [66]. Spectroflourimetric Methods: Spectrofluorimetric method was adopted for analysis of losartan in rabbit plasma. For this aim, a negative strong fluorescence was produced after dissolving of losartan in sulfurous acid solution. The mixture was measured at λem 410 nm using an excitation wavelength of λexc 248 nm [67]. Further, moreover spectroflourometric method was described for the determination of losartan in tablet dosage form by measuring a negative fluorescence of it upon dissolving drug in acidic medium. This fluorescence was measured at λem 400 nm using an excitation wavelength of λexc 247 nm [68].
Chromatographic Methods: HPLC-UV method was developed and validated for the simultaneous determination of hydrochlorothiazide and losartan in bulk and pharmaceutical formulation. The method was optimized selecting chromatographic conditions of 60: 40 acetonitrile : water, ACE3-C18 column (250 mm × 4.6 mm, 5 μm), 20 μl injection volume, flow rate of 1 ml/ min at ambient temperature, and 226 nm detection wavelength [69]. Simultaneous determination of losartan was performed by Kirtawade, et al. [70] on a RP C18 column using a mobile phase of acetonitrile : methanol : 25 Mm phosphate buffer (35:35:30, v/v/v) where pH was adjusted to 3.0 with phosphoric acid. The detection was set at 225 nm. Additionally, RP-HPLC method was developed for determination of losartan in pharmaceutical dosage form. The analysis was carried out using a Nucleodur 100 C18 column with UV detection at 210 nm. The mobile phase consisted of potassium dihydrogen phosphate buffer (pH 3.0) using diluted ortho phosphoric acid and acetonitrile (50:50, v/v) [71]. Vijayalakshmi and Dhanaraju [72] described RP-HPLC method with application of UV spectrophotometry (first-derivative, ratio-spectra first and second derivative) for determination of losartan potassium and hydrochlorothiazide in their binary mixtures. Methanol and phosphate buffer (pH 6.7) in the ratio of (80:20, v/v) were used as a mobile phase. Separation was performed on phenomenex C18 column with detection wavelength at 225 nm.
Another RP-HPLC method was developed for the determination of losartan potassium and perindopril erbumine in combined tablet dosage forms. The analysis was performed on a HiQSil-C18 W ODS column through a gradient elution system using acetonitrile : water in proportion of (50:50, v/v), pH 3.2. Drugs were detected at 210 nm [73]. RP-HPLC methods were developed for the determination of losartan and other drugs in their solid dosage form and in tablet dosage forms [74-79]. Chromatographic methods have been widely applied for determination of losartan potassium in pure form and in pharmaceutical formulations. These methods include RP-HPLC for the determination of losartan in tablets. As a column of BDS Hypersil C18 (250 × 4.6mm, 5μm) was used and acetonitrile : 0.2M phosphate buffer (pH: 3) at a proportion of (50:50, v/v) was the selected mobile phase. The detection of wavelength was set at 230 nm [80]. HPLC method was developed for the determination of losartan. Using a mixture of acetonitrile : 0.2M phosphate buffer (pH 3.0) (50:50, v/v) as mobile phase, a BDS Hypersil C18 column and UV detection at 230 nm [81]. HPTLC method was developed for the determination of losartan in pharmaceutical dosage form. The mobile phase was composed of a mixture of n-butanol : acetic acid : water (15: 5: 1, v/v/v) and the measurement was set at λmax 232 nm [82]. Losartan was assayed in its pharmaceutical formulations by RP-HPLC using a spherical monomeric C18 column. The mobile phase consisted of 0.01 M ammonium acetate buffer (pH 5.5) : acetonitrile (45:55, v/v) with detection wavelength at 240 nm [83]. HPLC method was developed for the determination of losartan potassium and amlodipine besilate in the presence of their acid, alkali, hydrolysis, photolysis, and oxidizing agent-induced degradation products. The chromatographic analysis was achieved by using Inertsil ODS-3 and buffer : acetonitrile (75:25, v/v) as a mobile phase [84].
Miscellaneous Methods: The cathodic stripping voltammetric behavior was studied on hanging mercury drop electrode to optimize an analytical method for determination of losartan in bulk and pharmaceutical dosage forms. The reaction was carried out in britton-robinson buffer solution at pH 7.0 [85].
Candesartan Scheme [3]
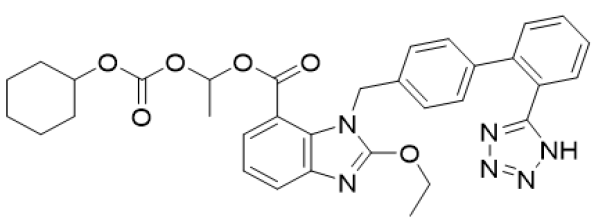
Scheme 3: Candesartan
a. Chemical Name: (1RS)-1-[[(Cyclohexyloxy) carbonyl] oxy] ethyl 2-ethoxy-1-[[2-(1 H-tetrazol-yl) biphenyl-4-yl] methyl]- 1H-benzimidazole.
b. Molecular Formula: C33H34N6O6.
c. Molecular Weight: 611.0 gm/mol.
d. Physical Properties: White or almost white powder, practically insoluble in water, freely soluble in methylene chloride and slightly soluble in anhydrous ethanol.
e. Melting Point: 183- 185 °C.
Pharmacological Action
Candesartan is an angiotensin II receptor blocker type 1 and used in treatment of various disorders like hypertension, heart failure, myocardial infarction, and diabetic nephropathy [86].
Official Method of Determination
Candesartan is official in B.P. 2013 [1] where it is determined by a potentiometric titration with 0.1 M perchloric acid in anhydrousacetic acid medium.
Literature Review
Spectrophotometric Methods: Stability-indicating spectrophotometric assay method for determination of candesartan cilexetil in presence of its alkaline degradation product. This method depended on using second derivative spectrophotometry at 291 nm [87]. The UV spectrophotometric method was used for the determination of candesartan; performed at λmax 212 nm by using methanol as a solvent [88]. The stabilityindicating spectrophotometric method was described for the determination of candesartan in bulk and pharmaceutical dosage form by using of first derivative spectrophotometry at 268 nm [89]. Five different hydrotropic agents like, potassium acetate, potassium citrate, sodium acetate, sodium citrate and urea were used for spectrophotometric determination of candesartan at λmax 280 nm [90]. Eglal, et al. [91] applied two dual wavelengths (method I) and mean centering of ratio spectra (MCR) (method II) for spectrophotometric determination of two combinations. The first combination consisted of hydrochlorothiazide and benazepril hydrochloride and the second consisted of hydrochlorothiazide and candesartan cilexetil in their bulk powder and tablets. PCR and PLS were used for determination of candesartan cilexetil and hydrochlorothiazide in their pharmaceutical preparations [92].
Candesartan cilexetil and atenolol were estimated in pure form and in pharmaceutical tablets where different blanks for each drug composed of methanol : distilled water (8:2) and 0.1M hydrochloric acid HCl were applied and scanned at specific wavelengths of 254 nm and of 224 nm for candesartan cilexetil and atenolol, respectively [93]. Candesartan reacted with DCQC and NQS in alcoholic medium forming colored complexes. These complexes were measured at λmax 550 nm and 480 nm, respectively [94]. The colored ion-pair complex was formed upon the reaction of BCG or BCP with candesartan in phosphate buffered solution. These complexes were measured at λmax 415, 405 nm with BCG, BCP, respectively [95].
Spectrofluorimetric Methods: Spectroflourimetric determination of candesartan was reported by direct measurement of fluorescence intensity. The emission and excitation wavelength were measured at 381 nm and 260 nm, respectively [96].
Chromatographic Methods: RP-HPLC method was described for determination of candesartan cilexetil in bulk and pharmaceutical dosage forms. Chromatographic separation was performed on Hypersil ODS C18 column. The mobile phase was a mixture of acetonitrile : 0.05 M KH2PO4 buffer in the ratio of (65:35, v/v) with detection wavelength at 256 nm [97]. Kamalakkannan, et al. [98] developed RP-HPLC method for the simultaneous estimation of candesartan cilexetil in bulk and in pharmaceutical dosage forms. Chromatographic separation was achieved on an Octa Decyl Silyl column with a mixture of phosphate buffer (pH 2.5) : acetonitrile (20:80, v/v) as mobile phase with detection wavelength at 215 nm. Revathi, et al. [99] described RP-HPLC method for validation of dissolution test for the quality control of candesartan cilexetil tablets by using 0.02M mono potassium phosphate : acetonitrile : triethylamine in the ratio of (40:60:0.2, v/v/v) as the mobile phase where pH was adjusted to 6 with phosphoric acid. RP-HPLC method was developed for the quantitative determination of candesartan. The chromatographic analysis was achieved by using a stainlesssteel column-Zorbax SB-CN. A mixture of 40 volumes of 10 Mm of sodium dihydrogen orthophosphate and 60 volumes acetonitrile was used as the mobile phase with detection wavelength at 210 nm [100]. Another RP-HPLC method was developed for quantification of candesartan.
The analysis was carried out using C18 intersil column with UV detection at 228 nm. The mobile phase consisted of acetonitrile : methanol (40:60 %, v/v, pH 6) [101]. Candesartan was determined in presence related impurities through HPLC method using Kromasil C18 reversed phase HPLC column and 0.01 M of ammonium acetate buffer 45% (pH 4.5) and 55% acetonitrile as a mobile phase. The detection was measured at 257 nm [102]. UPLC method was used for determination of candesartan cilexetil in bulk active pharmaceutical ingredient using a C18 stationary phase column and simple mobile phase combination in a gradient mode at 210 nm [103]. Candesartan was determined by RP-HPLC using a 0.02 M mono basic potassium phosphate buffer : actonitrile : triethylamine (40:60:0.2, v/v/v) as a mobile phase and pH was adjusted to 6.0 with orthophosphoric acid. The absorbance was measured at 254 nm [104]. Veeranjaneyulu, et al. [105] applied RP-HPLC method for the determination of candesartan cilexetil and hydrochlorothiazide in pharmaceutical dosage forms on hypersil BDS C column using phosphate buffer : acetonitrile (55:45, v/v) as a mobile phase (pH 4.6). The detection was carried out at wavelength 244 nm. Another RP-HPLC method was developed for determination of candesartan and amlodipine in pure form and combined solid dosage forms. The analysis which was based on using a reversed-phase column Phenomenex Luna, C18, (150 × 4.6 mm, 5 μm) with a mobile phase consisting of 0.1% orthophosphoric acid (pH 3.0) and acetonitrile in gradient programme. The UV detector was operated at 230 nm [106]. Another RP-HPLC method was developed for determination of candesartan in pure and pharmaceutical dosage forms.
The mobile phase consisted of a mixture of buffer (0.5% triethylamine) and methanol (50:50, v/v) and adjust the pH to 4.5 by using glacial acetic acid pumped through an Inertsil ODS-3V C18, 5 um columns. Peak was monitored by UV absorbance at 210 nm [107]. RP-HPLC method was developed for the determination of candesartan in bulk and tablet dosage forms. The analysis was performed on a Hypersill BDS-C8 (150 x 4.6mm, 5μm) using a degassed mixture of buffer and acetonitrile (40:60, v/v) as a mobile phase. Drug was detected at 210 nm [108]. Madhavi, et al. [109] developed HPLC method for the determination of candesartan cilexetil and hydrochlorothiazide. The chromatographic separation was performed on Silanol BDS C18 column (250 x 4.6 mm, 5 μm), a mobile phase consisting of water (pH adjusted to 2.8 with orthophosphoric acid) : acetonitrile (30:70 % v/v), with a flow rate 1 ml/min and the detection wavelength of 210 nm using photodiode array (PDA) detector.
Miscellaneous Methods: Electrochemical method was used for the determination of candesartan cilexetil on a glassy carbon electrode using a new sensor and continuous coulometric fast fourier transformation cyclic voltammetry. The new sensor designed was based on silicon carbide nanoparticles and graphene nanosheets hybrid mixed with ionic liquid [110].
Atenolol Scheme [4]
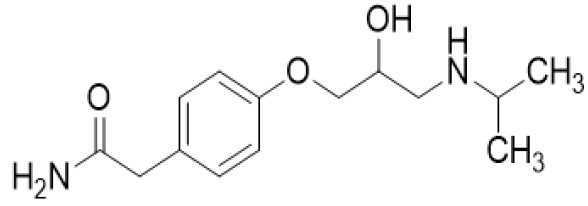
Scheme 4: Atenolol
a. Chemical Name: 2-{4-[(2RS)-2-Hydroxy-3-[(1-methylethyl) amino] propoxy] phenyl} acetamide.
b. Molecular Formula: C14H22N2O3.
c. Molecular Weight: 266.3 gm/mol.
d. Physical Properties: White or almost white powder, sparingly soluble in water, soluble in anhydrous ethanol, slightly soluble in methylene chloride, freely soluble in methanol.
e. Melting Point: 146 – 148 °C.
Pharmacological Action
Losartan is used in the treatment of hypertension and heart failure by inhibiting the rennin-angiotensin system [41]. It’s used to prevent occurring stroke in patients with hypertension [42] and it decreases an aortic-root enlargement so it’s better than atenolol and is considered a standard therapy to aortic-root dissection which is the main cause of death in Marfan’s syndrome [43].
Official Method of Determination
Losartan is an official in B.P. 2013 [1] where a potentiometric titration with perchloric acid in anhydrous-acetic acid medium is described.
Literature Review
Spectrophotometric Methods: Comparison of UV, second derivative spectrophotometric and HPLC methods were used for the determination of losartan in tablets [44] and also there was a comparison between first derivative spectrophotometry and HPLC methods that was used for the determination of losartan potassium in tablets [45]. Different colored charge-transfer complexes were formed as a result of reaction between losartan and TCQ, TNB, DDQ, p.chloranilic acid, TCE, TBQ, TCQ and TNF. These complexes were examined by UV-Vis, IR, 1H NMR techniques, and computational molecular modeling [46]. Alkaline potassium permanganate was used for oxidation of losartan potassium in pharmaceutical formulation and this reaction was measured spectrophotometrically at λmax 603 [47]. In addition, bromothymol blue was used as chromogen for spectrophotometric determination of losartan potassium at λmax 620 nm [48]. DDQ reacted with losartan potassium to give charge transfer complex at λmax 460 nm [49]. Chandra, et al. [50] described UV method for a comparative study of three brands of losartan potassium tablets. The detection was carried out at wavelength 205 nm and water: methanol was used as a solvent for the determination of losartan at λmax 275 nm [51]. Extractive spectrophotometric determination of losartan was developed using reagents; ferroin solution and methyl orange as ion-pairing agents (pH 7). The ion pair complex formed, which was extracted with chloroform, was measured at λmax 571 nm [52].
Sandeep and Marina [53] applied the Vierordt’s simultaneous equation method for the determination of losartan potassium in bulk and pharmaceutical dosage forms. Different spectrophotometric methods were developed for the determination of losartan potassium in combined dosage forms through dual wavelength and measurement of area under curve [54]. Two spectrophotometric methods were developed for the determination of losartan in tablet dosage form through Q-value analysis and simultaneous equation [55,56]. Spectrophotometric method was described for the determination of losartan in bulk and tablet dosage form by using sodium hydroxide [57] or methanol [58] as a solvent. The absorbance was measured at λmax 228 nm or 202 nm, respectively. On the other hand, ratio spectra derivative method was used for the estimation of losartan in combined dosage form. It showed maximum absorbance at 249 nm for losartan [59]. The first order UV-derivative spectrophotometry; applying zero-crossing method was developed for the determination of losartan in pharmaceutical formulations and wavelength of detection was set at 267 nm [60- 62]. The second order UV-derivative spectrophotometric method was described for the estimation of losartan in pharmaceutical forms and measuring absorbance at λmax 234 nm [63,64].
UV Spectrophotometric method was applied for the determination of losartan in tablet dosage forms. The method was based on an observation that the aqueous solution of losartan potassium exhibits an absorbance maximum at 205nm [65]. Spectrophotometric method had been developed for the quantitative estimation of losartan potassium in pharmaceutical formulation. The method depended on using double distilled water as a dissolving agent and measuring absorbance at λmax 205 nm [66].
Spectroflourimetric Methods: Spectrofluorimetric method was adopted for analysis of losartan in rabbit plasma. For this aim, a negative strong fluorescence was produced after dissolving of losartan in sulfurous acid solution. The mixture was measured at λem 410 nm using an excitation wavelength of λexc 248 nm [67]. Further, moreover spectroflourometric method was described for the determination of losartan in tablet dosage form by measuring a negative fluorescence of it upon dissolving drug in acidic medium. This fluorescence was measured at λem 400 nm using an excitation wavelength of λexc 247 nm [68].
Chromatographic Methods: HPLC-UV method was developed and validated for the simultaneous determination of hydrochlorothiazide and losartan in bulk and pharmaceutical formulation. The method was optimized selecting chromatographic conditions of 60: 40 acetonitrile : water, ACE3-C18 column (250 mm × 4.6 mm, 5 μm), 20 μl injection volume, flow rate of 1 ml/ min at ambient temperature, and 226 nm detection wavelength [69]. Simultaneous determination of losartan was performed by Kirtawade, et al. [70] on a RP C18 column using a mobile phase of acetonitrile : methanol : 25 Mm phosphate buffer (35:35:30, v/v/v) where pH was adjusted to 3.0 with phosphoric acid. The detection was set at 225 nm. Additionally, RP-HPLC method was developed for determination of losartan in pharmaceutical dosage form. The analysis was carried out using a Nucleodur 100 C18 column with UV detection at 210 nm. The mobile phase consisted of potassium dihydrogen phosphate buffer (pH 3.0) using diluted ortho phosphoric acid and acetonitrile (50:50, v/v) [71]. Vijayalakshmi and Dhanaraju [72] described RP-HPLC method with application of UV spectrophotometry (first-derivative, ratio-spectra first and second derivative) for determination of losartan potassium and hydrochlorothiazide in their binary mixtures. Methanol and phosphate buffer (pH 6.7) in the ratio of (80:20, v/v) were used as a mobile phase. Separation was performed on phenomenex C18 column with detection wavelength at 225 nm.
Another RP-HPLC method was developed for the determination of losartan potassium and perindopril erbumine in combined tablet dosage forms. The analysis was performed on a HiQSil-C18 W ODS column through a gradient elution system using acetonitrile : water in proportion of (50:50, v/v), pH 3.2. Drugs were detected at 210 nm [73]. RP-HPLC methods were developed for the determination of losartan and other drugs in their solid dosage form and in tablet dosage forms [74-79]. Chromatographic methods have been widely applied for determination of losartan potassium in pure form and in pharmaceutical formulations. These methods include RP-HPLC for the determination of losartan in tablets. As a column of BDS Hypersil C18 (250 × 4.6mm, 5μm) was used and acetonitrile : 0.2M phosphate buffer (pH: 3) at a proportion of (50:50, v/v) was the selected mobile phase. The detection of wavelength was set at 230 nm [80]. HPLC method was developed for the determination of losartan. Using a mixture of acetonitrile : 0.2M phosphate buffer (pH 3.0) (50:50, v/v) as mobile phase, a BDS Hypersil C18 column and UV detection at 230 nm [81]. HPTLC method was developed for the determination of losartan in pharmaceutical dosage form. The mobile phase was composed of a mixture of n-butanol : acetic acid : water (15: 5: 1, v/v/v) and the measurement was set at λmax 232 nm [82]. Losartan was assayed in its pharmaceutical formulations by RP-HPLC using a spherical monomeric C18 column. The mobile phase consisted of 0.01 M ammonium acetate buffer (pH 5.5) : acetonitrile (45:55, v/v) with detection wavelength at 240 nm [83].
HPLC method was developed for the determination of losartan potassium and amlodipine besilate in the presence of their acid, alkali, hydrolysis, photolysis, and oxidizing agent-induced degradation products. The chromatographic analysis was achieved by using Inertsil ODS-3 and buffer : acetonitrile (75:25, v/v) as a mobile phase [84].
Miscellaneous Methods: The cathodic stripping voltammetric behavior was studied on hanging mercury drop electrode to optimize an analytical method for determination of losartan in bulk and pharmaceutical dosage forms. The reaction was carried out in britton-robinson buffer solution at pH 7.0 [85].
Candesartan Scheme [3]
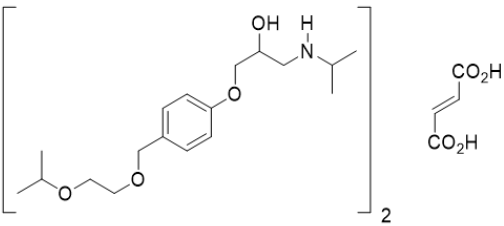
Scheme 5: Bisoprolol Fumarate
a. Chemical Name: (2RS)-1-{4-[(2-(isopropoxyethoxy) methyl] phenoxy}-3-[isopropyl amino] propan -2-ol fumarate.
b. Molecular Formula: C40H66N2O12.
c. Molecular Weight: 767 gm/mol.
d. Physical Properties: White or almost white powder, slightly hygroscopic powder, very soluble in water, freely soluble in methanol.
e. Melting Point: 100 °C.
Pharmacological Action
Candesartan is an angiotensin II receptor blocker type 1 and used in treatment of various disorders like hypertension, heart failure, myocardial infarction, and diabetic nephropathy [86].
Official Method of Determination
Candesartan is official in B.P. 2013 [1] where it is determined by a potentiometric titration with 0.1 M perchloric acid in anhydrousacetic acid medium.
Literature Review
Spectrophotometric Methods: Stability-indicating spectrophotometric assay method for determination of candesartan cilexetil in presence of its alkaline degradation product. This method depended on using second derivative spectrophotometry at 291 nm [87]. The UV spectrophotometric method was used for the determination of candesartan; performed at λmax 212 nm by using methanol as a solvent [88]. The stabilityindicating spectrophotometric method was described for the determination of candesartan in bulk and pharmaceutical dosage form by using of first derivative spectrophotometry at 268 nm [89]. Five different hydrotropic agents like, potassium acetate, potassium citrate, sodium acetate, sodium citrate and urea were used for spectrophotometric determination of candesartan at λmax 280 nm [90]. Eglal, et al. [91] applied two dual wavelengths (method I) and mean centering of ratio spectra (MCR) (method II) for spectrophotometric determination of two combinations. The first combination consisted of hydrochlorothiazide and benazepril hydrochloride and the second consisted of hydrochlorothiazide and candesartan cilexetil in their bulk powder and tablets. PCR and PLS were used for determination of candesartan cilexetil and hydrochlorothiazide in their pharmaceutical preparations [92].
Candesartan cilexetil and atenolol were estimated in pure form and in pharmaceutical tablets where different blanks for each drug composed of methanol : distilled water (8:2) and 0.1M hydrochloric acid HCl were applied and scanned at specific wavelengths of 254 nm and of 224 nm for candesartan cilexetil and atenolol, respectively [93]. Candesartan reacted with DCQC and NQS in alcoholic medium forming colored complexes. These complexes were measured at λmax 550 nm and 480 nm, respectively [94]. The colored ion-pair complex was formed upon the reaction of BCG or BCP with candesartan in phosphate buffered solution. These complexes were measured at λmax 415, 405 nm with BCG, BCP, respectively [95].
Spectrofluorimetric Methods: Spectroflourimetric determination of candesartan was reported by direct measurement of fluorescence intensity. The emission and excitation wavelength were measured at 381 nm and 260 nm, respectively [96].
Chromatographic Methods: RP-HPLC method was described for determination of candesartan cilexetil in bulk and pharmaceutical dosage forms. Chromatographic separation was performed on Hypersil ODS C18 column. The mobile phase was a mixture of acetonitrile : 0.05 M KH2PO4 buffer in the ratio of (65:35, v/v) with detection wavelength at 256 nm [97]. Kamalakkannan, et al. [98] developed RP-HPLC method for the simultaneous estimation of candesartan cilexetil in bulk and in pharmaceutical dosage forms. Chromatographic separation was achieved on an Octa Decyl Silyl column with a mixture of phosphate buffer (pH 2.5) : acetonitrile (20:80, v/v) as mobile phase with detection wavelength at 215 nm. Revathi, et al. [99] described RP-HPLC method for validation of dissolution test for the quality control of candesartan cilexetil tablets by using 0.02M mono potassium phosphate : acetonitrile : triethylamine in the ratio of (40:60:0.2, v/v/v) as the mobile phase where pH was adjusted to 6 with phosphoric acid. RP-HPLC method was developed for the quantitative determination of candesartan. The chromatographic analysis was achieved by using a stainlesssteel column-Zorbax SB-CN. A mixture of 40 volumes of 10 Mm of sodium dihydrogen orthophosphate and 60 volumes acetonitrile was used as the mobile phase with detection wavelength at 210 nm [100]. Another RP-HPLC method was developed for quantification of candesartan.
The analysis was carried out using C18 intersil column with UV detection at 228 nm. The mobile phase consisted of acetonitrile : methanol (40:60 %, v/v, pH 6) [101]. Candesartan was determined in presence related impurities through HPLC method using Kromasil C18 reversed phase HPLC column and 0.01 M of ammonium acetate buffer 45% (pH 4.5) and 55% acetonitrile as a mobile phase. The detection was measured at 257 nm [102]. UPLC method was used for determination of candesartan cilexetil in bulk active pharmaceutical ingredient using a C18 stationary phase column and simple mobile phase combination in a gradient mode at 210 nm [103]. Candesartan was determined by RP-HPLC using a 0.02 M mono basic potassium phosphate buffer : actonitrile : triethylamine (40:60:0.2, v/v/v) as a mobile phase and pH was adjusted to 6.0 with orthophosphoric acid. The absorbance was measured at 254 nm [104]. Veeranjaneyulu, et al. [105] applied RP-HPLC method for the determination of candesartan cilexetil and hydrochlorothiazide in pharmaceutical dosage forms on hypersil BDS C column using phosphate buffer : acetonitrile (55:45, v/v) as a mobile phase (pH 4.6). The detection was carried out at wavelength 244 nm. Another RP-HPLC method was developed for determination of candesartan and amlodipine in pure form and combined solid dosage forms.
The analysis which was based on using a reversed-phase column Phenomenex Luna, C18, (150 × 4.6 mm, 5 μm) with a mobile phase consisting of 0.1% orthophosphoric acid (pH 3.0) and acetonitrile in gradient programme. The UV detector was operated at 230 nm [106]. Another RP-HPLC method was developed for determination of candesartan in pure and pharmaceutical dosage forms.
The mobile phase consisted of a mixture of buffer (0.5% triethylamine) and methanol (50:50, v/v) and adjust the pH to 4.5 by using glacial acetic acid pumped through an Inertsil ODS-3V C18, 5 um columns. Peak was monitored by UV absorbance at 210 nm [107]. RP-HPLC method was developed for the determination of candesartan in bulk and tablet dosage forms. The analysis was performed on a Hypersill BDS-C8 (150 x 4.6mm, 5μm) using a degassed mixture of buffer and acetonitrile (40:60, v/v) as a mobile phase. Drug was detected at 210 nm [108]. Madhavi, et al. [109] developed HPLC method for the determination of candesartan cilexetil and hydrochlorothiazide. The chromatographic separation was performed on Silanol BDS C18 column (250 x 4.6 mm, 5 μm), a mobile phase consisting of water (pH adjusted to 2.8 with orthophosphoric acid) : acetonitrile (30:70 % v/v), with a flow rate 1 ml/min and the detection wavelength of 210 nm using photodiode array (PDA) detector.
Miscellaneous Methods: Electrochemical method was used for the determination of candesartan cilexetil on a glassy carbon electrode using a new sensor and continuous coulometric fast fourier transformation cyclic voltammetry. The new sensor designed was based on silicon carbide nanoparticles and graphene nanosheets hybrid mixed with ionic liquid [110].
Atenolol Scheme [4]
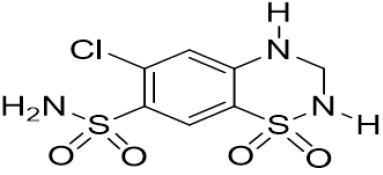
Scheme 6: Hydrochlorothiazide
a. Chemical Name: 6-Chloro-3, 4-dihydro-2H-1, 2, 4-benzothiadiazine -7-sulfonamide 1, 1-dioxide.
b. Molecular Formula: C7H8ClN3O4S.
c. Molecular Weight: 297.7 gm/mol.
d. Physical Properties: White or almost white, crystalline powder, very slightly soluble in water, soluble in acetone, sparingly soluble in ethanol. It dissolves in dilute solutions of alkali hydroxides.
e. Melting Point: 273-275 °C.
Pharmacological Action
Atenolol is b-blocker [111] used for the treatment of hypertension without compelling indications [112].
Official Method of Determination
Atenolol is official in B.P. 2013 [1] which describes a potentiometric titration with perchloric acid in anhydrous-acetic acid medium.
Literature Review
Spectrophotometric Methods: Pawar, et al. [113] described UV spectrophotometric method for the determination of atenolol and indapamide in a binary mixture. In this method, the signals were measured at 225 nm and 240 nm, respectively after dissolving atenolol and indapamide in methanol. In addition, two methods were used for the determination of atenolol in combined tablet dosage form. The first method was based on absorbance ratio at 232 nm (iso-absorptive point) whereas, the second method was depending on the first order derivative spectroscopy using 214 nm (zero cross for atenolol) [114]. Atenolol and chlorthalidone were dissolved in methanol followed by further dilutions with methanol then absorbance maxima were found to be 225 nm and 284 nm, respectively [115]. Another UV spectrophotometric method was used for determination of atenolol and atorvastatin in tablet dosage forms. The method was based upon dissolving drugs in methanol then determination of λmax of both drugs at 225 nm and 241 nm, respectively [116]. Edebi, et al. [117] described titrimetric and UV spectrophotometric methods for the qualitative and quantitative determination of atenolol in tablet form. The titrimetric method was based on the use of non-aqueous acetous perchlorate while the spectrophotometric method was based on evaluation in organic solvent medium of binary λmax of 275 nm and 282 nm.
The colored complexes were formed between atenolol and DDQ, DNP and TNP, were measured at λmax 590 nm, 420 nm and 420 nm, respectively [118]. Kinetic spectrophotometric method for the determination of atenolol in bulk form and tablet was described. The method was based on measuring the decrease in absorbance of cerium at λ 360 nm [119]. DDQ was utilized for determination of atenolol forming charge transfer complex with maximum absorbance at λmax 405 nm [120]. Zero crossing method was used for the simultaneous determination of atenolol in combined dosage form. The absorbance was measured at λmax 275 nm [121]. UV-spectrophotometric method was applied for the estimation of atenolol in its tablet dosage forms. The absorbance was measured at λmax 273.2 nm in ammonium acetate solution [122]. The marketed combination of atenolol and amlodipine besylate was dissolved in 0.1N HCl. The spectra of amlodipine and atenolol exhibited λ of 239 nm and 228 nm, respectively [123]. Second order UV derivative spectrophotometric method for the determination of atenolol was used for quantification at 245 nm [124]. Moreover, Kudige and Nagaraju [125] described a spectrophotometric method for the determination of atenolol in bulk drug and in tablets. Atenolol was dissolved in 0.1N sodium hydroxide then measuring the absorbance at 273 nm.
Afaf, et al. [126] described several methods for simultaneous determination of atenolol and chlorthalidone. The first method depended on first derivative of the ratio’s spectra by measurements of the amplitudes at 235 nm for atenolol and (236, 249 nm) for chlorthalidone. The second method depended on measuring the absorbance at the isosbestic point at 283.5 nm for the total concentration of both drugs where the concentration of chlorthalidone was determined by direct spectrophotometric method at λ 250 nm in the presence of atenolol, while the concentration of atenolol is calculated by subtraction. The third method was factorized absorptivity method at which both drugs are determined at more than one isosbestic point (265, 284 nm).
Spectroflourimetric Methods: Spectroflourometric method was described for the determination of atenolol in tablet dosage form by measuring its strong fluorescence upon dissolving drug in 0.1N sodium hydroxide in boiling water bath. The resulting product exhibited fluorescence at λem 302 nm when excited at λ exc 278 nm [127].
Chromatographic Methods: Bhaskara, et al. [128] described HPLC method for determination of atenolol in pharmaceutical formulation; using Atlantis C column, 20 mM ammonium acetate : methanol (60:40, v/v) as mobile phase with UV detection at 225 nm. GC-MS was used for determination of atenolol in human urine. Atenolol and metoprolol (as internal standard) were extracted from human urine by liquid liquid extraction then derivatization reaction occurred between these extracts and MSTFA (as a chromagenic derivatization reagent) to increase sensitivity [129]. Walaa El-Alfy, et al. [130] developed a simple RP-HPLC-PDA method for determination of atenolol and trimetazidine in human urine and tablets. Analytes were separated on a Caltrex BI column (125× 4.0 mm, 5 μm) with 25mM potassium dihydrogen phosphate pH 3.3, methanol, and acetonitrile mobile phases. The PDA detector was operated at 210 nm for trimetazidine and 225 nm for atenolol and the flow rate was 1.0 mL/ min. RP-HPLC method was developed for the determination of atenolol and hydrochlorothiazide in tablet form using Xterra ODS C18 column, water, and methanol (50:50, v/v) as mobile phase and UV detector at 230nm [131].
HPTLC method was used for simultaneous determination of atenolol and losartan potassium in tablet dosage form. The separation was performed on precoated Silica Gel 60 F-254 aluminum sheets by using mobile phase consisting of ethyl acetate : methanol : 1,4 dioxane : ammonia (10:2:1:2, v/v/v/v) with UV detection at 225 nm [132]. Furthermore, another RP-HPLC and UV spectroscopy were used for determination of atenolol and lercanidipine hydrochloride in bulk and tablet dosage forms [133]. Ravi, et al. [134] described UPLC method for determination of atenolol in tablet dosage forms. This method was performed on Zorbax-C18 column using mobile phase consisting of buffer (sodium- 1-heptane sulfonate and anhydrous dibasic sodium phosphate) and methanol in the ratio (70:30, v/v) with UV detection at 226 nm.
Miscellaneous Methods: An ion-selective electrode was used for determination of atenolol in pharmaceutical preparation. This electrode consisted of membrane containing atenolol-phospho tungstate complex and di-octyl phthalate (as a plasticizer) [135]. Diffuse reflectance spectroscopy was used for the determination of atenolol in pharmaceutical formulations. In this method, the drug reacted with p-chloranil producing a colored compound with maximum absorption at λmax 550 nm [136]. Iontophoresis method was used to raise transdermal permation of atenolol from films [137]. Prashanth, et al. [138] described non-aqueous titrimetric method for the determination of atenolol in bulk and pharmaceutical formulations. The method was based on the titration of the drug in glacial acetic acid with acetous perchloric acid to the visual end point using crystal violet as indicator or to the potentiometric end point using a modified glass electrode-SCE system. Oxidimetric reagents, like potassium bromate and acid dyes, methyl orange and indigo carmine were used for titrimetric and spectrophotometric determination of atenolol and propranolol hydrochloride in pharmaceutical dosage form [139].
Determination of atenolol and propranolol in pharmaceutical formulations using square-wave voltammetry and a cathodically pretreated borondoped diamond electrode was described. These electroanalytical determinations of propranolol or atenolol were carried out in 0.1 mol L-1 H2SO4 or 0.5 mol L-1 NaNO3 (pH 1.0, adjusted with concentrated HNO3), respectively [140].
Bisoprolol Fumarate Scheme [5]
Pharmacological Action
Bisoprolol is b-blocker and is used in treatment of hypertension and heart failure [141,142].
Official Method of Determination
Bisoprolol is official in B.P. 2013 [1] which describes a potentiometric titration with perchloric acid in anhydrous-acetic acid medium.
Literature Review
Spectrophotometric Methods: Spectrophotometric determination of bisoprolol fumarate and amlodipine besylate in 10% methanol was based on using simultaneous equation method that had been carried out at two wavelengths 222 nm and 365 nm, respectively [143]. Hydrotropy solubilization agents were used for simultaneous spectrophotometric determination of bisoprolol fumarate in tablets with maximum absorbance at wavelength 271nm [144]. The formation of an ion pair complex between bisoprolol and methyl orange in acid medium was used as spectrophotometric method for determination of bisoprolol. The produced complex was measured at 427 nm [145]. Two spectrophotometric methods were used for the determination of propafenone HCl, naftidrofuryl oxalate and bisoprolol hemifumarate in pure form and pharmaceutical preparations. The first method depended on formation of ion -pair complexes between the drugs and two dyes namely tropeolin 000 or chromazurol S while the second method depended on formation of ternary complex between the drugs and the binary inorganic complex mo (v)-thiocyanate then extraction with methylene chloride [146]. Fadel and Anas [147] applied the bivariate calibration method for determination of hydrochlorothiazide and bisoprolol fumarate in pharmaceutical tablets at new λmax 273.5 nm and 293 nm, respectively.
Visible spectrophotometric method was developed for the determination of bisoprolol based on the formation of blue colored chromogen when drug reacted with ferric chloride and pottasium ferricynide. This blue colored complex was measured at 770 nm [148]. A very simple, accurate and rapid spectrophotometric method was developed for estimation of bisoprolol fumarate and levobunolol hydrochloride using excess ceric ammonium sulphate in sulphuric acid medium as oxidant. The unreacted Ce+4 have been determined using two different dyes indigo carmine and methyl orange. The colored solutions of indigo carmine and methyl orange have maximum absorbance at 610 nm and 504 nm respectively [149].
Spectrofluorimetric Methods: Abd El-Kawy, et al. [150] described spectrofluorimetric determination of bisoprolol fumarate and valsartan in spiked plasma and tablets .The fluorescent product was formed in 0.1N H2SO4 with excitation wavelength at 227 nm and emission wavelength at 298 nm.
Chromatographic Methods: Li, et al. [151] developed LC– ESI-MS method for the determination of bisoprolol in human plasma, using metoprolol as internal standard. After alkalization with sodium hydroxide, the samples were extracted with ethyl acetate then separated on ZORBAX SB-C18 column with a mobile phase of 10 mM ammonium acetate buffer containing 0.1% formic acid–methanol (32:68, v/v). Fluorescence HPLC method was developed for determination of bisoprolol in human plasma, based on derivatization with NBD-Cl in borate buffer at pH 9.5. The resulting a fluorescent product was determined on a C18 reversed phase column by using mixture of methanol : water (70:30, v/v) with fluorescence detector at λexc 458 nm /λem 525 nm [152]. Stability indicating liquid chromatography was performed by Madhusudhanareddy, et al. [153] in which bisoprolol fumarate was separated on a C18 column with ammonium dihydrogen orthophosphate (buffer) (pH 5.6) : acetonitrile (75:25, v/v) as a mobile phase and detected using a diode array at 226 nm. A simple, specific, and precise RP-HPLC method was developed and validated for simultaneous determination of bisoprolol fumarate and rosuvastatin calcium in new formulated tablets.
The developed RP-HPLC method depended on chromatographic separation using C18 column (150 × 4.6 mm, 5 μm) with mobile phase consisting of acetonitrile and 0.05 aqueous solution of orthophosphoric acid at the ratio of 65:35 % (v:v) with a flow rate of 1 mL/min and UV detection at 230 nm [154]. HPTLC method was used for simultaneous determination of bisoprolol and hydrochlorothiazide in pharmaceutical dosage forms. The separation was performed on precoated Silica Gel 60 F-254 aluminum sheets by using mobile phase consisted of ethyl acetate : methanol : ammonia (10:0.5:0.5, v/v/v) at 225nm [155].
Miscellaneous Methods: Differential pulse voltammetric method was described for the determination of bisoprolol in pharmaceutical formulations at glassy carbon electrode. The reaction was carried out in 0.5 M phosphate buffer solution at pH 7.2 [156]. Jingwu, et al. [157] described CZE coupled with tris (2, 2′-bipyridyl) ruthenium (II)-based end-column ECL for determination of bisoprolol after separation from metoprolol and other co-existed materials in tablets and urine samples. Deshmukh, et al. [158] described direct compression method using sodium starch glycolate, croscarmellose sodium and crospovidone as super disintegrants in different concentrations for fast dissolving tablets of bisoprolol fumarate. Direct compression and wet granulation were used with different proportion and types of diluents and different binder types for preparation of bisoprolol fumarate tablets [159].
Hydrochlorothiazide Scheme [6]
Pharmacological Action
Hydrochlorothiazide is a thiazide diuretic and is considered the most commonly used as antihypertensive drug [160]. It decreases blood pressure by direct arterial dilation [161]. It is used alone or in a combination with other antihypertensive agents such as b-blockers and rennin angiotensin system blockers [162].
Official Method of Determination
Hydrochlorothiazide is official in B.P. 2013 [1] where it was determined by chromatographic system.
Literature Review
Spectrophotometric Methods: Spectrophotometric method was based on using chlorpheniramine maleate as a hydrotropic solubilizing agent to solubilize hydrochlorothiazide from tablets then analysis was carried out at 317 nm [163]. Nikam, et al. [164] described first order derivative UV spectrophotometric method for estimation of valsartan, amlodipine besylate and hydrochlorothiazide in combined dosage form .The absorbance of three drugs was measured at 245, 265 and 279 nm, respectively. UV spectrophotometric method was used for the determination of amlodipine besylate, valsartan and hydrochlorothiazide in bulk and combined tablet form by dissolving three drugs in methanol followed by the further dilutions. Absorbance was measured at 239 nm, 250 nm and 272 nm, respectively [165]. Patel, et al. [166] described Q-absorbance equation spectrophotometric method for determination of hydrochlorothiazide and candesartan cilexetil in tablet dosage forms. UV-spectrophotometric and ion pair chromatographic method had been used for determination valsartan and hydrochlrothiazide in tablet dosage forms [167]. Continuous wavelet transformation was developed for spectrophotometric determination of hydrochlorothiazide in tablet dosage form. The experimental results obtained from this method were compared with those obtained from derivative spectrophotometry [168].
Abd El kawy, et al. [169] described ratio subtraction method for resolving the overlapping spectra of two binary mixtures: moexipril hydrochloride with hydrochlorothiazide and fosinopril with hydrochlorothiazide in pharmaceutical preparations. The signals were measured at 205 nm and 209 nm respectively. Simultaneous equation, absorption ratio and first order derivative spectroscopy methods had been used for spectrophotometric determination of amlodipine besylate and hydrochlorothiazide in tablet form [170]. Simultaneous equation spectrophotometric method was used for estimation of hydrochlorthiazide in bulk and pharmaceutical formulations. The absorbance was measured at λmax of 271nm for hydrochlorothiazide after dissolving in methanol [171]. First order derivative spectrophotometric method was described for determination of nebivolol and hydrochlorothiazide in tablets .The absorbance was measured at 270 nm and 282 nm for nebivolol and hydrochlorothiazide, respectively [172]. Oxidation of hydrochlorothiazide by ammonium metavanadate formed complex which was measured spectrophotometrically at λmax 365 nm [173].
Another UV spectrophotometric method for determination of hydrochlorothiazide and valsartan in solid dosage forms was described. This method was based on dissolving drugs in methanol then absorbance was measured at 271 nm for hydrochlorothiazide [174]. Two spectrophotometric methods were presented for estimation of atenolol and hydrochlorothiazide in combined dosage form. The first method was area under curve and the absorbance was measured between 221 to 231 nm and 265 to 275 nm for atenolol and hydrochlorothiazide, respectively. The second method was second order derivative spectrophotometry, and the absorbance was measured at 226 nm and 270 nm for atenolol and hydrochlorothiazide, respectively [175].
Different solvents such as distilled water and 0.01 N NaOH were used for the determination of hydrochlorothiazide in tablet dosage form and measuring the absorbance at λmax 272 nm and between 200 nm and 400 nm in case of distilled water and 0.01N NaOH, resbectively [176]. Two methods were described for spectrophotometric determination of hydrochlorothiazide in bulk and in pharmaceutical dosage forms through simultaneous equation method and absorption correction method then the absorbance was measured at 271.2 nm and 316.4 nm, respectively [177]. Absorbance ratio spectrophotometric method was developed for the determination of hydrochlorothiazide in bulk and pharmaceutical formulations. Hydrochlorothiazide had shown an isoabsorptive point at 287 nm in methanol [178].
UV spectrophotometric method was applied for estimation of hydrochlorothiazide in bulk drug by using 6M urea and 10% citric acid to achieve hydrotropic solubilization of drug in pharmaceutical dosage forms. The absorbance was measured at 272 nm [179].
Chromatographic Methods: RP-HPLC was used for the determination of enalapril maleate and hydrochlorothiazide in tablet formulation. The separation was performed on a Supelcosil LC8 column with a mobile phase consisting of 3.0 mM tetra butyl ammonium hydrogen sulfate in acetonitrile/ water/ triethylamine (14: 85.6: 6.4, v/v/v) adjusted to pH 4.1 by glacial acetic acid and detection was carried out at 220 nm [180]. HPLC method was developed for the determination of irbesartan and hydrochlorothiazide in combined dosage form. The chromatographic analysis was achieved by using a supelcosil C18 column and methanol : tetra hydrofuran : acetate buffer (47:10:43, v/v/v) as a mobile phase adjusted to pH 6.5. The detection was carried out at wavelength 271 nm [181]. Dhanalakshmi , et al. [182] described RP-HPLC method for the determination of telmisartan and hydrochlorothiazide in tablet formulation. The separation was carried out on column C8 with mixture of ammonium phosphate buffer and acetonitrile (60:40, v/v) as a mobile phase. The detection was carried at 270 nm. HPTLC method was used for simultaneous determination of olmesartan medoxomil and hydrochrolothiazide in pharmaceutical preparations. The separation was performed on silica gel 60 F254 plates using toluene : methanol : ethylacetate : acetone (2.5:1:0.5:2, v/v/v/v) as the mobile phase. The analysis was carried out at 258 nm [183].
UPLC was developed for the determination of valsartan and hydrochorothiazide in combined dosage forms using kromasil eternity C18 column and triethylamine buffer (0.1 % v/v) (pH 3.0 adjusted with o-phosphoric acid) and methanol (75:25, v/v) as a mobile phase. The detection wavelength was measured at 225 nm [184]. RP-HPLC method for the determination of hydralazine and hydrochlorothiazide in bulk and pharmaceutical dosage forms. The separation was carried out on Inertsil ODS C18 using mixture of 0.01M potassium dihydrogen orthophosphate buffer and acetonitrile (60:40, v/v) as a mobile phase, pH was adjusted to 4.8 with orthophosphoric acid. Drugs were detected at 217 nm [185].
Miscellaneous Methods: Diffuse reflectance spectroscopy was performed for the determination of hydrochlorothiazide in pharmaceutical formulations by the reaction between hydrochlorothiazide and p-dimethyl amino cinnamaldehyde (PDAC) in acid medium to form colored compound [186]. The electrochemical oxidation behavior of hydrochlorothiazide was studied on a glassy carbon as a working electrode to optimize an analytical method for the determination of hydrochlorothiazide in pharmaceutical formulations and biological fluids. The reaction was carried out in britton-robinson buffer solution at pH3 [187]. Three chemometric calibrations; principal component regression (PCR), partial least squares (PLS) and artificial neural network (ANN) were used for determination telmisartan and hydrochlorotiazide in the commercial pharmaceuticals [188].
References
- (2013) The British pharmacopoeia. HM Stationary Office, London, UK.
- Christy L, Ralph H (2001) Agiotensin receptor blockers (ARB). Southern Medical Journal, 94(11).
- Adams MA, Trudeau L (2000) Irbesartan: Review of pharmacology and comparative properties. Canadian Journal Clinical Pharmacology 7(1): 22-31.
- Bernard W (2001) A review of irbesartan in antihypertensive therapy comparison with other antihypertensive agent. Current Therapeutic Research 62(7): 505-523.
- Chunyi W, Cunyun M, Xianglu R, Tingting F, Xuhui H, et al. (2015) Irbesartan can improve blood lipid and the kidney function of diabetic nephropathy. Discovery Medicine 20(108): 67-77.
- Watanabe R, Suzuki JI, Wakayama K, Kumagai H, Ikeda Y, et al. (2015) Angiotensin II receptor blocker irbesartan attenuates cardiac dysfunction induced by myocardial infarction in the presence of renal failure. Hypertension Research 39(4): 237-244.
- Wang XJ, Hu W, Zhang TY, Mao YY, Liu NN, et al. (2015) Irbesartan, an FDA approved drug for hypertension and diabetic nephropathy, is a potent inhibitor for Hepatitis B virus entry by disturbing Na(+)-dependent taurocholate cotransporting polypeptide activity. Antiviral Research 120: 140-146.
- Senda A, Mukai Y, Toda T, Hayakawa T, Yamashita M, et al. (2015) Effects of angiotensin II receptor blockers on metabolism of arachidonic acid via cyp2c8. Biological and Pharmaceutical Bulletin 38(12): 1975-1979.
- Vandana SN, Pranita PD, Laxman AK (2016) Anti-fibrotic effect of irbesartan via attenuation of endoplasmic reticulum stress in isoprenaline-induced myocardial fibrosis. International Journal of Pharmacology and Clinical Sciences 24(4): 76-82.
- Patel KR, Patel SA, Darji VC, Sonpal RN (2011) Simultaneous spectrophotometric estimation of irbesartan and hydrochlorothiazide in tablets. International Research Journal of Pharmacy 2(3): 202-207.
- Tuljarani G, Gowrishankar D, Shireesha M, Satyanarayana B (2011) Spectrophotometric method for determination of angiotensin-II receptor antagonist in bulk and pharmaceutical dosage forms. International Journal of Pharmacy and Pharmaceutical Sciences 4(1): 198-202.
- Arshiya F, Sayaji R, Venkateshwarlu G (2011) Spectrophotometric determination of drugs using 2, 3 dichloro 5, 6 dicyano P-benzoquinone as analytical reagent. International Journal of ChemTech Research 3(4): 1769-1780.
- Darwish I, wani T, Khalil N, Bakheit A (2012) Novel 96- microwell spectrometric assay with high throughput for determination of irbesartan in its tablets for determination of irbesartan in its tablets. Digest Journal of Nanomaterials and Biostructures 7(2): 415-421.
- Dhanawade P, Kane R (2012) Derivative spectrophotometric method for estimation of Irbesartan in bulk drug and dosage form. International Journal of Research in Pharmaceutical and Biomedical Science 3(3): 1300-1304.
- Vemugunta R, Anupama B (2012) Assay of irbesartan by extractive spectrophotometry. International Journal of Pharmaceutical Chemical and Biological Sciences 2(4): 529-531.
- Anupama B, Abhinav K, Suri N, Surendra A (2012) UV Spectrophotometric method for irbesartan. International Journal of Researchin Pharmacy and Chemistry 2(1): 20-21.
- Laxmi B, Jay KC, Prabhat P (2013) Method development and validation estimation of irbesartan in bulk drug and pharmaceutical dosage. Journal of Drug Delivery & Therapeutics 3(6): 87-90.
- Srinath N, Anil KA, Rama DB, Krishna PV (2013) Estimation of irbesartan in bulk and dosage forms by new simple uv spectrophotometry using hydrotropic technique. Pharmaceutica Analytica Acta 4(8): 1-3.
- Mohamed M, Salwa R, Samiha A, Niveen A (2013) Validated non-Extractive spectrophotometric methods for determination of some angiotensin II receptor antagonists. Asian Journal Pharmaceutical Analysis 3(1): 03-08.
- Amit A, Anita S, Suman M, Asati k (2014) Quantitative analysis method development and validation for irbesartan in bulk drug by ultraviolet spectroscopy. Journal of Advanced Pharmacy Education & Research 4(1): 101-105.
- Ashour S, Bayram R (2019) Selective and validated kinetic spectrophotometric method for the determination of irbesartan in pure and pharmaceutical formulations. Annales Pharmaceutiques Françaises 77(2): 101-111.
- Lakshmi S, Lakshmi K (2014) H-point standard addition method for simultaneous spectrophotometric determination of irbesartan, hydrochlorothiazide and telmisartan in tablets. International Journal of Research in Pharmacy and Chemistry 4(2): 373-380.
- Farouk M, Abd EL-Aziz O, Hemdan A, Shehata M (2011) Simple Novel spectrophotometric and spectrofluorimetric methods for determination of some anti-hypertensive drugs. Journal of American Science 7(1): 300-312.
- Ramzia I, Hanaa M, Waleed A (2011) Septrofluorometric, Spectrophotometric and LC Determination of Irbesartan. Journal of Chemical and Pharmaceutical Research 3(4): 722-733.
- Afaf A, Gamal H, Hisham A, Eman A (2016) Spectrofluorimetric and spectrophotometric determination of irbesartan and bisoprolol hemifumarate independently in their tablets. UK Journal of Pharmaceutical and Biosciences 4(2): 43-52.
- Aniruddha R, Swati C, Bhanudas S, Pradeep D, Kunal D, et al. (2010) Development and validation of a RP-HPLC-PDA method for simultaneous estimation of hydrochlorothiazide and irbesartan. Der Pharma Chemica 2(4): 148-156.
- Krystyna C, Aleksander P (2011) Identification and determination of selected angiotensin II receptor antagonist group drugs by HPLC method. Acta Poloniae Pharmaceutica - Drug Research 68(6): 831-837.
- Rosangluaia S, Malarkodi V (2011) Validated HPTLC method for simultaneous of irbesartan and hydrochlorothiazide in tablet dosage forms. Der Pharma Chemica 3(5): 310-317.
- Mhaske R, Sahasrabudhe S, Mhaske A (2012) RP-HPLC method for simultaneous determination of irbesartan, losartan, hydrochlorothiazide, and chlorthalidone-application to commercially available drug products. International Journal of Pharmaceutical Sciences and Research 3(4): 1116-1123.
- Raja B, Potru H, Bantu R (2012) RP-HPLC Method for the simultaneous estimation of irbesartan and hydrochlorothiazide in pharmaceutical dosage form. International Research Journal of Pharmaceutical and Applied Sciences 2(3): 29-38.
- Hany M, Abdullah A, Lobna M, Magda M (2013) Quantitative determination of three angiotensin-II-receptor antagonists in presence of hydrochlorothiazide by RP-HPLC in their tablet preparations. Iranian Journal of Pharmaceutical Research 12(4): 635-643.
- Ramesh B, Elizabeth Y, Dhanalaxmi K, Nagarjuna R (2013) Method development and validation of irbesartan and hydrochlorothiazide by RP-HPLC in bulk and pharmaceutical dosage form. International Journal of Pharmacy 3(3): 521-526.
- Mohammed E, Mohamed A, Dina T, Dalia R, Saadia M (2013) Simultaneous determination of irbesartan and hydrochlorothiazide in pharmaceutical preparations and spiked human plasma using microemulsion liquid chromatography. International Journal of Advances in Pharmaceutical Research 4(7): 1944-1959.
- Zаrеh MM, Sааd MZ, Hаssаn WS, Еlhеnnаwу MЕ, Soltan MK, et al. (2020) Grаdiеnt HPLC Mеthоd fоr Simultаnеоus Dеtеrminаtiоn оf Еight Sаrtаn аnd Stаtin Drugs in Thеir Purе аnd Dоsаgе Fоrms. Pharmaceuticals 13(2): 32.
- Reem Y, Adnan H, Ahmad H (2014) Determination and separation of valsartan, losartan and irbesartan in bulk and pharmaceutical formulation by RP-HPLC. International Journal of Pharmaceutical Sciences Review and Research 27(2): 169-172.
- Seema Z, Tanveer A (2014) New UPLC–MS/MS method for simultaneous determination of irbesartan and hydrochlorothiazide in human plasma. Journal of the Iranian Chemical Society 11(6).
- Gandla K, Seshagiri R, Rajendra K (2015) New stability indicating validated RP-HPLC method for simultaneous estimation of irbesartan and atorvastatin incombined tablet dosage forms. American journal of pharmaceutical research 5(4): 381-390.
- Vinod K, Rajeev J, Shilpi A, Ritesh M, Ashish D (2011) Electrochemical determination of antihypertensive drug irbesartan in pharmaceuticals. Analytical Biochemistry 410(2): 266-271.
- Hanaa S, Mohamed M, Allia D (2011) Voltammetry of irbesartan drug in pharmaceutical formulations and human blood: Quantification and pharmacokinetic studies. Journal of the Brazilian Chemical Society 22(2): 239-247.
- Shrikant H, Minakshi V (2012) Novel and validated titrimetric method for determination of selected angiotensin-II-receptor antagonists in pharmaceutical preparations and its comparison with UV spectrophotometric determination. Journal of Pharmaceutical Analysis 2(6): 470-477.
- Peter KS, Salah D, Theodore P (1995) Pharmacology of losartan, an angiotensin II receptor antagonist in animal models of hypertension. Journal of Hypertension 13(1): S15-S21.
- Javier D (2006) Review of the molecular pharmacology of losartan and its possible relevance to stroke prevention in patients with hypertension. Clinical Therapeutics 28(6): 832-848.
- Ronald V, Harry C, Lynn A, Anji T, Timothy J, et al. (2014) Atenolol versus Losartan in Children and Young Adults with Marfan's Syndrome. New England Journal of Medicine 371: 2061-2071.
- Sevgi T, Serap S (2004) Comparison of UV- and second derivative spectrophotometric and high-performance liquid chromatographic methods for the determination of losartan in tablets. Turkish Journal Pharmaceutical Sciences 1(3): 165-175.
- Mehdi A, Maryam K, Freshteh K, Mehdi B (2004) A Comparative study of first-derivative spectrophotometry and high-performance liquid chromatography applied to the determination of losartan potassium in tablets. Chemical and Pharmaceutical Bulletin 52(10): 1166-1170.
- Ibrahim AD (2005) Analytical study for the charge-transfer complexes of losartan potassium. Analytica Chimica Acta 549: 212-220.
- Nafisur R, Masoom R, Syed N (2006) Development, and validation of kinetic spectrophotometric method for the determination of losartan potassium in pure and commercial tablets. Journal of the Chinese Chemical Society 53(3): 735-743.
- Latheeshjlal L, Parthiban P, Alagarsamy M, Vaidhya M, Rama M (2010) Spectrophotometric determination of lorsartan potassium and its dosage form by bromothymol blue and phosphate buffer. E-Journal of Chemistry 7(1): 320-324.
- Ibrahim AD, Ashraf MM, Abdul Rahman AA (2010) Novel analytical approach for reducing the consumption of organic solvents in the charge transfer-based spectrophotometric analysis of losartan International Journal Research of Pharmaceutical Science 1(4): 391-395.
- Chandra D, Desireddy R, Jitendrakumar P, Suresh N, Sravankumar G, et al. (2012) A comparative study of three brands of losartan potassium tablets by UV Spectrophotometry. International Journal of Chemical and Pharmaceutical Sciences 3(3): 76-78.
- Gajbhiye A, Dwivedi N (2012) Simultaneous estimation of Losartan and Atenolol by UV Spectrophotometric Method. Current Trends in Technology and Science 1(3): 141-145.
- Nikita DP, Anandkumari DC (2013) Extractive spectrophotometric method for simultaneous determination of losartan potassium and atenolol in bulk and in pharmaceutical dosage form. International Journal of PharmTech Research 5(2): 629-640.
- Sandeep S, Marina K (2014) Spectrophotometric quantitative estimation of atenolol and losartan potassium in bulk drugs and pharmaceutical dosage form. World Journal of Pharmacy and Pharmaceutical Sciences 3(7): 1026-1033.
- Chaitali T, Jyoti D, Pawar P (2014) Simultaneous estimation and validation of losartan potassium and hydrochlorothiazide in bulk and tablet dosage form by using different spectrophotometric method. Der Pharma Chemica 6(2): 24-30.
- Priyanka R, Sachin U, Dhabale P, Burade K (2009) Simultaneous UV Spectrophotometric method for estimation of losartan potssium and amlodipine besylate in tablet dosage form. Asian Journal Research Chemistry 2(1): 183-187.
- Pragati KB, Prafulla KS, Abhinov T (2011) Simple spectrophotometric methods for simultaneous determination of losartan potassium and atorvastatin calcium in combined dosage forms. International Journal of Chemical and pharmaceutical sciences 4(3): 127-131.
- Kapil K, Nidhi G, Shailendra P (2010) Development of new UV spectrophotometric method for estimation of losartan potasium in bulk and tablet dosage Form. International Journal of Medical Sciences 2(2): 110-111.
- Ramya G, Puranik S, Kumar G, Sridha K (2012) Simultaneous estimation of amlodipine and losartan by UV-method in bulk drug and tablet dosage formulation. Archives Applied Science Research 4(5): 2206-2212.
- Surjyanarayan M, Jignesh G, Rashmita M, Rajesh K (2014) Ratio spectra derivative method development and validation for the drug combination of losartan potassium and chlorthalidone. Agenesis Journal Pharmagene 1(4): 40-46.
- Mehdi A, Maryam K, Mehdi B, Hassan J (2004) Derivative spectrophotometric method for determination of losartan in pharmaceutical formulations. Iranian Journal of Pharmacology &Therapeutics 3(1): 21-25.
- Tarkase K, Suryawanshi S, Joshi R (2012) Simultaneous derivative spectrophotometric determination and validation of losartan potassium in pharmaceutical dosage forms. International Journal of Pharmaceutical Sciences Review and Research 13(2): 31-35.
- Laxmi B, Pranita K (2012) Analytical Method development and validation for the estimation of losartan by first order derivatives pectroscopy. Journal of Pharmaceutical Science and Technology 4(4): 917-923.
- Rao P, Venugopal V, Anil K, Rajesh B, Prasad G, et al. (2011) Quantitative estimation of losartan potassium in pharmaceutical dosage forms by UV spectrophotometry. International Journal of Research in Pharmacy and Chemistry 1(3): 295-302.
- Tangri P, Singh P, Mukhopadhyay S, Tangri S (2012) Development and validation of uv spectrophotometric method for the estimation of losartan bulk drug and pharmaceutical formulation. International Research Journal of Pharmacy 3(5): 391-393.
- Subbarao J, Venkateswara R, Vidyadhara S, Venkateswara R (2012) UV spectrophotometic validation for identification and determination of losartan potassium in tablets. International Journal of Pharmacy& Technology 4(1): 4137-4143.
- Ajit CJ, Sadhana RS, Imran T, Santosh DB (2016) Method development of antihypertensive agent using official dissolution media. International Journal of Pharmaceutical Research & Allied Sciences 5(1): 58-64.
- Fatma D, Mehmet E, Yucel K (2015) A new spectrofluorimetric method for determination of losartan potassium in rabbit plasma and its application to pharmacokinetic study. The Journal of Biological and Chemical Luminescence 30(1): 53-59.
- Taher M, Asadollahzadeh H, Fazelirad H (2015) A simple spectrofluorimetric method for the determination of losartan in some tablet dosage forms. Journal of Applied Spectroscopy 82(5): 842-844.
- Nidhal MSM, Hasan RA, Hassan MH (2019) Method development and validation of simultaneous determination of hydrochlorothiazide and losartan in tablet dosage form by RP-HPLC. International Journal of Pharmaceutical Sciences and Research 10(1): 227-231.
- Kirtawade R, Salve P, Kulkarni A, Dhabale P (2010) RP-HPLC method for simultaneous estimation of losartan potassium and atenolol in tablet formulation. An International Journal of Pharmaceutical Sciences 1(2): 50-56.
- Havaldar FH, Vairal DL (2010) Simultaneous estimation of atenolol, hydrochlorothiazide losartan and valsartan in the pharmaceutical dosage form. International Journal of Pharmacy & Life Sciences 1(5): 282-289.
- Vijayalakshmi R, Dhanaraju M (2011) Application of UV spectrophotometry and RP-HPLC techniques for the determination of binary mixtures of losartan k and hydrochlorthiazide. Journal of Pharmacy Research 4(7): 2310-2312.
- Oza C, Prajapati J, Vyas A, Mehta P (2011) RP-HPLC method for the determination of losartan potassium and perindopril erbumine in combined tablet dosage form. International Journal of Pharma and Bio Sciences 2(1): 709-715.
- Nitin S, Lalitha K (2014) Validated method development for simultaneous estimation of losartan potasium and chlorthalidone in tablet dosage form by RP-HPLC method. World Journal of Pharmaceutical Research 3(7): 410-419.
- Sindu Y, Ajitha A, Uma M (2014) Analytical method development and validation for simultaneous estimation of losartan potassium and enalapril maleate in tablet dosage form by RP-HPLC. International Journal of Pharmacy 4(4): 314-319.
- Khan MR, Shaikh A, Thaker A (2012) Simultaneous determination and method development for assay of losartan potassium and hydrochlorothiazide drugs in solid dosage form by RP-HPLC. Indian Journal of Pharmaceutical Science & Research 2(1): 42-45.
- Aruna G, Chanukya M, Reddy M (2012) Simultaneous estimation of losartan potassium and hydrochlorothiazide drugs in solid dosage form by RP-HPLC. International Journal of Medicinal Chemistry & Analysis 2(1): 57-61.
- Sharma R, Khanna S, Mishra G (2013) RP-HPLC method for simultaneous estimation of atenolol, hydrochlorothiazide and losarton in tablet dosage form. Chemical Science Transactions 2(S1): S1-S6.
- Ganipisetty L, Dachinamoorthy D, Seshagiri R (2014) A sensitive RP-HPLC method development and validation for the simultaneous estimation of losartan potassium and hydrochlorothiazide. International Journal for Pharmaceutical Research Scholars 3(2): 408-416.
- Sundar R, Afsar S, Sarathchandiran I, Srinivasan G (2013) Development of a validated high-performance liquid chromatography method for the simultaneous determination of losartan potassium and atenolol in tablets. International Journal Pharmaceutical Biomedical Science 4(3): 100-105.
- Magda MI, Maha AH, Abd El-Aziz E, Mohamed AA (2012) Rapid and sensitive HPLC method for simultaneous estimation of atorvastatin, and losartan hydrochlorothiazide and quantitative application to polypill based synthetic ternary mixture. Journal of Chemical and Pharmaceutical Research 4(11): 4737- 4742.
- Mariusz Stolarczyk, Anna Maslanka, Anna Apola, Jan krzek (2013) Determination of losartan potassium, quinapril hydrochloride and hydrochlorothiazide in pharmaceutical preparations using derivative spectrophotometry and chromatographic- densitometric method. Acta Poloniae Pharmaceutica-Drug Research 70 (6): 967-967.
- Hani MH, Abdullah AE, Lobna MA, Magda MK (2014) Quantitative determination of amlodipine besylate, losartan Potassium, valsartan and atorvastatin calcium by HPLC in their pharmaceutical formulations. Pharmaceutical Analytical Acta Journal 5(5): 1-5.
- Korhale R, Chaudhari P, Neeta W, Pravin M, Ruby S, et al. (2014) Analytical assay method validation and forced degradation behavior of s (-) amlodipine besilate and losartan potassium from fixed dose combination. International Journal of Universal Pharmacy and Bio Sciences 3(4): 280-295.
- Habib I, Weshahy S, Toubar S, El-Alamin M (2008) Cathodic stripping voltammetric determination of losartan in bulk and pharmaceutical products. Portugaliae Electrochimica Acta 26(4): 315-324.
- Shantanu B, Lohit B, Devi L (2013) An explicit review on quantitative estimation of candesartan cilexetil employing various analytical techniques. Pharmaceutica Analytica Acta 4(6): 1-7.
- Nawal AA, Wedad AA, Amina ME (2011) Spectrophotometric determination of candesartan cilexetil in presence of its alkaline induced degradation product. Asian Journal of Chemistry 23(4): 1696-1700.
- Padmalatha H, Vidyasagar G (2011) Quantitative estimation of candesartan by UV spectrophotometry. International Journal of Pharmacy& Technology 3(2): 2653-2658.
- Kalyani G, Vaishnav Y, Deshmukh S, Sahu R (2013) Stability indicating method development and validation of candesartan in bulk and pharmaceutical dosage form by derivative spectrophotometric method. International Journal of Pharmamedix India 1(2): 222-232.
- Yogesh P, Agrawal M, Gupta A (2013) Spectrophotometric estimation of candesartan cilexetil by using different hydrotropic agents. International Journal of Pharmaceutical Sciences Letters 3(2): 190-194.
- Eglal AA, Ibrahim AN, Hala EZ, Mohammed ED (2013) Simultaneous determination of some antihypertension drugs in their binary mixtures by simple spectrophotometric methods. Asian Journal of Biomedical and Pharmaceutical Sciences 3(25): 5-12.
- Wedad AA, Hany WD, Nawal AA, Amina MA (2013) Application of PCR and PLS methods for the simultaneous determination of candesartan cilexetil and hydrochlorothiazide in their pharmaceutical preparations. Digest Journal of Nanomaterials and Biostructures 8(3): 1253-1262.
- Sura L, Rajwan AA, Abdulbari MM (2020) Spectrophotometric Determination of Candesartan Cilexetil and Atenolol in Pure and Pharmaceutical Forms. International Journal of Pharmaceutical Research 12(3).
- Muralikrishna C, Rambabu C (2015) Determination of candesartan cilexetil in bulk and pharmaceutical dosage forms by visible spectrophotometric methods. International Journal of Science and Research 4(8): 492-495.
- Alaa SA, Hanna MS, Gamal HR, Inass SK (2015) Sensitive spectrophotometric and conductometric methods for determination of candesartan using bromocresol green and bromocresol purple by ion-pair complex formation. Journal of Chemistry and Materials Research 2(2): 42-48.
- Amir A, Hanan F (2010) Determination of candesartan cilexetil in tablets by spectrofluorimetry. International Journal of Pharmaceutical Sciences Review and Research 4(1): 60-63.
- Ganesh A, Kandikonda S, Saikumar B, Rasapally R, Santhosh K (2010) RP- HPLC method development and validation of candesartan cilexetil in bulk and their pharmaceutical dosage forms. International Journal of Pharmaceutical Sciences and Research 1(12): 191-196.
- Kamalakkannan V, Puratchikody A, Masilamani K, Saraswathy T (2011) Analytical method development and validation for candesartan cilexetil as bulk drug and in pharmaceutical dosage forms by HPLC. Scholars Research Library 3(3): 286-296.
- Revathi R, Ethiraj T, Jhansi L, Ganeshan V (2011) Development and validation of a dissolution test for candesartan cilexetil in tablet forms using reverse phase – high performance liquid chromatography. Journal of Pharmaceutical Education and Research 2(2): 71-77.
- Naga D, Putta R, Salahuddin M, Shanta K (2012) Candesartan cilexetil analytical method development and validation studies by reverse phase hplc technique. International Journal of pharmaceutical Frontier Research 2(3): 36-43.
- Manisha P, Sailesh J, Ashish L, Pramod G (2012) Method development and validation of candesartan cilexetil by RP-HPLC. International Journal of Research in Pharmaceutical and Biomedical Sciences 3(3): 1227-1230.
- Katiyar M, Kushwaha D, Shukla R (2012) Specific and stability indicating assay method of cadesartan cilexetil in presence of process and degradation impurities. International Journal of Pharmaceutical Innovations 2(5): 1-10.
- Gunda S, Kakumani K, Gangaram V, Vishnu P, Mukkanti KA (2012) Stability indicating UPLC method for candesartan in bulk drug samples. American Journal of Analytical Chemistry 3: 704-709.
- Revathi R, Saravanan V, Ethiraj T, Jhansi L (2013) RP-HPLC analysis for quantitation of candesartan cilexetil in solid dosage forms. Asian Journal Pharmaceutical Analysis 3(4): 115-118.
- Veeranjaneyulu D, Aneesha A, Nandakishore A (2013) Stability indicating RP-HPLC method for the simultaneous determination of candesartan cilexetil and hydrochlorothiazide in bulk and dosage forms. Indian Journal of Research in Pharmacy and Biotechnology 1(5): 720-724.
- Kalaichelvi R, Srilakshmi G, Jayachandran E (2013) Gradient RP-HPLC method for simultaneous estimation of amlodipine and candesartan in tablets. International Journal of Universal Pharmacy and Life Sciences 3(6): 15-22.
- Syeda K, Vidya S, Nagalakshmi, Snehalatha R (2014) Development and validation of RP-HPLC method for estimation of candesartan from tablet dosage form. World Journal of Pharmacy and Pharmaceutical Sciences 3(4): 781-786.
- Lakshmi S, Naga S, Renuka K, Vinusha K (2014) Method development and validation for the estimation of candesartan cilexitil in bulk and tablet dosage forms by RP-HPLC. International Journal of Inventions in Pharmaceutical Sciences 2(1): 617-621.
- Madhavi K, Navamani M, Prasanthi C (2017) Simple analytical method for the simultaneous estimation of hydrochlorothiazide and candesartan by RP-HPLC. International Journal of Applied Pharmaceutics 9(6): 34-38.
- Norouzi P, Pirali H, Ganjali M (2013) Candesartan cilexetil determination by electrode modified with hybrid film of ionic liquid- graphene nanosheets-silicon carbide nanoparticle using continuous coulometric fft cyclic voltammetry. International Journal of Electrochemical Science 8(2): 2023-2033.
- Shelke OS, Sable KS, Neharkar VS, Mathdevru BV (2012) Development and validation of a UV spectrophotometric method for the simultaneous determination of nifedipine and atenolol in combine dosage form. International Research Journal of Pharmacy 3(4): 360-364.
- Laura MK, Nadia AK (2014) Atenolol vs Nonatenolol β-Blockers for the treatment of hypertension: A meta-analysis. Canadian Journal of Cardiology 30(5): S47-S53.
- Pawar P, Gaikwad P, Bankar V, Pawar S (2010) Development and validation of UV-spectrophotometric method for simultaneous estimation of atenolol and indapamide in bulk and tablet dosage form. International Journal of Pharmacy &Technology 2(4): 876-885.
- Behera A (2010) Simultaneous spectrophotometric estimation of atenolol and hydrochlorothiazide in tablet dosage forms. International Journal of ChemTech Research 2(4): 1901-1906.
- Akiful H, Nivedita G, Prashanth K, Pradeep K, Hasan A, et al. (2012) Simultaneous estimation of atenolol and chlorthalidone as bulk and in tablet dosage form using UV- Spectrophotometry. IOSR Journal of Pharmacy and Biological Sciences 1(4): 20-23.
- Dey S, Sarkar S, Malakar J, Ghosh A, Gangopadhyay A, et al. (2012) Spectrophotometric method for simultane ous determination of atenolol and atorvastatin in tablet dosage forms. International Journal of pharmaceutical and biomedical research 3(1): 40-43.
- Edebi V, Benjamin E, Joffa P (2012) Simple, sensitive and reproducible acetous perchlorate and spectrophotometric determination of atenolol in tablet dosage form. Journal of pharmaceutical sciences and research 4(10): 1933-1938.
- Kudige N, Kanakapura B (2012) Simple, sensitive and selective spectrophotometric methods for the determination of atenolol in pharmaceuticals through charge transfer complex formation reaction. Acta Poloniae Pharmaceutica- Drug Research 69(2): 213-223.
- Rashmi A, Anand G (2012) Kinetic spectrophotometric determination of atenolol in perchloric acid medium. International Journal of pharmacy and Pharmaceutical Sciences 4(2): 350-353.
- Nagaraja SK, Chakravarthi IE (2013) A UV-Visible Spectrophotometric determination of atenolol in pharmaceutical formulations. International Journal Scientific Research 2(3): 31-32.
- Lalitha K, Kiranjyothi P, Padma B (2013) UV spectrophotometric method development and validation for the determination of atenolol and losartan potassium by Q-analysis. International Bulletin of Drug Research 3(4): 54-62.
- Lalitha G, Salomip P, Ravindra R (2013) Development of an analytical method and its validation for the analysis of atenolol in tablet dosage form by UV- Spectrophotometry. International Journal of Pharmacy and Pharmaceutical Sciences 5(2): 197-199.
- Pawar P, Mane B, Sumit M, Vipul VT (2013) Simultaneous estimation of amlodipine besylate and atenolol in combined dosage form by Vierodt’s method using U.V. spectroscopy. Der Pharma Chemica 5(2): 97-102.
- Hiral B, Bhavna A, Shraddha P (2013) Development and validation of second order derivative spectrophotometric method for simultaneous estimation of atenolol and nifedipine in combined dosage form. International Journal of Pharmaceutical Sciences and Research 4(10): 3884-3888.
- Kudige N, Nagaraju S (2014) UV- Spectrophotometric Assay of atenolol in pharmaceuticals and study of its forced degradation. Asian Journal of Biochemical and Pharmaceutical Research 4(1): 240-253.
- Afaf A, Hanaa M, Magda M, Basma E (2015) Derivative ratio, isosbestic point, factorized absorptivity and bivariate spectrophotometric determination of atenolol and chlorthalidone. International Journal of Chemical and Biological Sciences 5(1): 19-33.
- Patel S, Patel C, Rathod I, Suhagia B, Patel M (2005) Spectrofluorimetric method for the determination of atenolol in tablet dosage forms. Indian Journal of Pharmaceutical Science 67(2): 224-226.
- Bhaskara B, Anil K, Anil K (2011) A facile and rapid HPLC method for determination of atenolol in pharmaceutical formulations. Asian Journal of Applied Sciences 4(3): 306-313.
- Bilal Y, Sakir A (2011) Determination of atenolol in human urine by Gas Chromatography–Mass Spectrometry method. Journal of Chromatographic Science 49: 365-369.
- El-Alfy W, Ismaiel OA, El-Mammli MY, Shalaby A (2019) Determination of Atenolol and Trimetazidine in Pharmaceutical Tablets and Human Urine Using a High-Performance Liquid Chromatography-Photo Diode Array Detection Method. International Journal of Analytical Chemistry p. 1-8.
- Medaharitha, Ramadevi B (2012) Method development and validation for the simultaneous estimation of atenolol and hydrochlorthiaz in tablet dosage form by RP-HPLC method. An International Journal of Advances in Pharmaceutical Sciences 3(3-4): 204-210.
- Swati M, Celina N, Leena S (2013) Development and validation of a new sensitive method for the quantitative analysis of atenolol-losartan potassium in a tablet dosage form by using HPTLC. Scholars Academic Journal of Pharmacy 2(6): 464-467.
- Mahendra S, Ravindra K, Chetan S (2014) Development and validation of lercanidipine hydrochloride and atenolol by using RP-HPLC and UV spectroscopy. Indian Journal of Pharmacy and Pharmacology 1(1): 38-41.
- Ravi K, Krishna R, Sasikiran G (2014) Development and validation of UPLC method for determination of atenolol in tablets form. World Journal of Pharmacy and Pharmaceutical Sciences 3(9): 808-816.
- Nabil SN, Shahbaz AM, Mutaz AA (2007) Preparation and characterization of an atenolol selective electrode based on a PVC matrix membrane. Turkish Journal Chemistry 31: 75-82.
- Gotardo M, Sequinel R, Pezza L, Pezza H (2008) Determination of atenolol in pharmaceutical formulations by diffuse reflectance spectroscopy. Ecl Quím São Paulo 33(4): 7-12.
- Ozge I, Muge K, Nurayari Tamer B (2008) In vitro and in vivo transdermal studies of atenolol using iontophoresis. Acta Poloniae Pharmaceutica ñ Drug Research 65(1): 29-36.
- Prashanth KN, Basavaiah K, Raghu MS, Vinay KB (2012) Determination of atenolol and its preparations by acid-base titration in non-aqueous medium. Der Pharmacia Lettre 4(5): 1534-1540.
- Bagheri A, Mesgarzadeh I (2012) Titrimetric and spectrophotometric methods for the determination of β-blockers in pharmaceutical dosage form. Journal of Applied Chemical Research 6(2): 53-64.
- Elen R, Roberta A, Romeu C, Orlando F (2010) Square-wave voltammetric determination of propranolol and atenolol in pharmaceuticals using a boron-doped diamond electrode. Talanta 81: 1418-1424.
- Tevfik F, Fethi K, Ata K, Mehmet U (2006) Bisoprolol improve echocardiographic parameters of left ventricular diastolic function in patients with systemic hypertension. Cardiology 106(3): 127-131.
- Gavin JK, Keating GM (2006) Bisoprolol, A review of its use in chronic heart failure. Drugs 62: 2677-2696.
- Kakde R, Kotak A, Chaudhary N, Kale D (2008) Spectrophotometric method for simultaneous estimation of amlodipine besylate and bisoprolol fumarate in pharmaceutical preparations. Research Journal Pharmacy and Technology 1(4): 513-515.
- Smita S, Mukesh C (2010) Novel method for spectrophotometric analysis of simultaneous estimation of bisoprolol fumarate tablet formulations using hydrotropy solubilization agents. Journal of Optoelectronics and Biomedical Materials 2(4): 223-225.
- ALina D, Andreea M, Vasile D (2012) Spectrophotometric determination of bisoprolol using methyl orange as reagent. Farmacia 60(5): 634-641.
- Magda A, Hisham A, Mervat H, Yassmin S (2013) Utility of complexation reaction for the determination of some cardiovascular drugs. Asian Journal of pharmaceutical and Clinical Research 6(1): 92-102.
- Fadel W, Anas L (2013) Application of the bivariate calibration for simultaneous determination oh hydrochlorothiazide/enalapri maleate and hydrochlorothiazide/bisoprolol fumarate in drug tablets. International Journal of Chemistry 5(2): 29-37.
- Smita T, Dattatrya B, Payal B (2013) Visible spectrophotometric method for estimation of bisoprolol from its bulk and tablet formulation. Asian Journal of Pharmaceutical and Clinical Research 6(4): 103-105.
- Elshaprawy DS, FE Mohammed, Elgendy K (2020) Spectrophotometric Determination of Bisoprolol Fumarate and Levobunolol Hydrochloride in Bulk and Pharmaceutical Forms. Eurasian Journal of Analytical Chemistry 15(1): emEJAC-00379.
- Abdel-Kawy M, Amer S, Lotfy H, Zaazaa H (2006) Spectrofluorimetric determination of bisoprolol fumarate and valsartan in tablets and spiked plasma. Egyptian Journal of Pharmaceutical Sciences 47: 107-116.
- Li D, Xia Z, Xiaofeng G, Qinxin S, Jianchang H, et al. (2007) LC–ESI-MS method for the determination of bisoprolol in human plasma. Journal of Pharmaceutical and Biomedical Analysis 44(2): 520-525.
- Sevgi T, Zeynep A (2012) An HPLC method for the determination of bisoprolol in human plasma and its application to a pharmacokinetic study. Journal of Chromatographic Scienc 50: 615-619.
- Madhusudhanareddy I, Bhagavan R, Rajendra P (2012) Validated and stability indicating liquid chromatography method for quantification of bisoprolol fumarate in tablet dosage form. International Journal Pharmacy 2(1): 64-70.
- Zaki MM, Abdelwahab NS, Ali AA, Sharkawi SMZ (2019) Simultaneous determination of bisoprolol fumarate and rosuvastatin calcium in a new combined formulation by validated RP-HPLC. European Journal of Chemistry 10(1): 52-56.
- Savita S, Janhavi R (2013) Simultaneous HPTLC analysis of bisoprolol fumarate and hydrochlorthiazide in pharmaceutical dosage form. International Journal of Pharmacy and Pharmaceutical Sciences 5(2): 286-290.
- Rajendra N, Anuradha T, Neeta B, Sunita B (2008) Voltammetric determination of bisoprolol fumarate in pharmaceutical formulations and urine using single-wall carbon nanotubes modified glassy carbon electrode. Electrochimica Acta 53(6): 2802-2808.
- Jingwu W, Xiaojun Z, Fangfang P, Xiaoxia W, Nianjun Y (2009) Tris (2,2′-bipyridyl) ruthenium(ii)- bisoprolol- based electrochemiluminescence coupled with capillary zone electrophoresis. Electrochimica Acta 54(8): 2379-2384.
- Deshmukh N, Thenge R, Mahajan N (2012) Formulation, and evaluation of bisoprolol fumarate fast dissolving tablet by direct compression techniques. International Journal of Pharmaceutical Research and Bioscience 1(5): 364-378.
- Heba A, Alaa A (2013) Effect of different diluent and binder types on the preparation of bisoprolol fumarates as tablet dosage form. Iraqi Journal Pharmaceutical Science 22(1): 32-39.
- Abhichandani V, Faruqui A (2014) Evaluation of triple drug combination (telmisartan, amlodipine and hydrochlorothiazide) in the management of Pacific Journal of Medical Sciences 12(2): 3-11.
- Jones, Bartlett (2015) Nurse's Grug Handbook pp. 570-572.
- Danielle C, Sherry M (2015) Duiretics for hypertension: Hydrochlorothiazide or chlorthalidone. Clevel and Clinic Journa of Medicine 82(8): 527-533.
- Maheshwari R, Bishnoi S, Kumar D (2008) Spectrophotometric analysis of hydrochlorothiazide tablets using chlorpheniramine maleate as hydrotropic solubilizing agent. Asian Journal of Chemistry 20(8): 6594-6596.
- Nikam M, Harshad D, Aniket A, Kondawar M (2010) Imultaneou estimation of valsartan, amlodipine besylate and hydrochlorothiazide by first order derivative UV spectrophotometric method. International Journal of Pharmacy&Technology 2(3): 642-650.
- Jothieswari D, Anandakumar K, Vijaya S, Vijayakumar B, Priya D, et al. (2010) A validated UV spectrophotometric method for the simultaneous estimation of amlodipine besylate, valsartan hydrochlorothiazide in bulk and in combined tablet dosage form. Journal of Pharmaceutical and Biomedical Sciences 5(13): 1-5.
- Patel J, Dave J, Patel C, Patel D (2010) Q-Analysis spectrophotometric methods for estimation of candesartan cilexetil and hydrochlorothiazide in tablet dosage form. Journal of Chemical and Pharmaceutical Research 2(3): 10-14.
- Bhatia N, Bhatia M, Choudhari P, Ingale K (2010) Development and validation of spectrophotometric and ion pair chromatographic technique for estimation of valsartan and hydrochlorothiazide. Journal of Pharmaceutical Research and Health Care 2(1): 2-14.
- Mahmoud R, Khadijeh M, Atieh J (2010) Simultaneous spectrophotometric determination of triamterene and hydrochlorothiazide in Triamterene-H tablets using continuous wavelet transformation. Journal of Applied Chemical Researche 4(14): 61-67.
- Abd El Kawy M, El Gindy A, Hegazy M, Shokry E (2010) Novel spectrophotometric method for simultaneous determination of two binary mixtures containing hydrochlorothiazide by ratio subtraction. Journal of Applied Sciences Research 6(8): 918-926.
- Vijaya V, Vrushali T, Vrushali K, Dhole S (2011) Spectrophotometric simultaneous determination of amlodipine besylate and hydrochlorothiazide in combined tablet dosage form by simultaneous equation, absorption ratio and first order derivative spectroscopy methods. International Journal of Chemistry Research 2(1): 7-10.
- Patel R, Patel J (2011) Simultaneous equation spectrophotometric method for the estimation of nebivolol HCl and hydrochlorthiazide in their combined dosage form. International Journal of Pharmaceutical and Applied Sciences 2(1): 6-10.
- Satish A Patel, Hemant M Patel (2011) Simultaneous determination of nebivolol and hydrochlorthiazide in tablets by derivative spectrophotometry. American Journal of Pharmtech Research 1(4): 421-429.
- Neeti N, Sandeep K, Ramprakash A, Jagdish P, Archna P (2011) A new spectrophotometric method for the determination of hydrochlorothiazide based on the redox reaction. Journal of Materials Science and Engineering 1: 725-730.
- Sunil S, Ajit K, Hemendra G (2011) Simultaneous estimation of valsartan and hydrochlorothiazide in solid dosage form using UV spectroscopy. Bulletin of Pharmaceutical Research 1( 3): 10-12.
- Manzoor A, Nadeem J, Satishkumar S (2012) Simultaneous estimation of atenolol and hydrochlorothiazide in combined dosage form by UV-spectrophotometric methods. Acta Chimical Pharmaceutical Indica 2(3): 134-142.
- Hapse SA, Wagh VS, Kadaskar PT, Dokhe MD, Shirsath AS (2012) Spectrophotometric estimation and validation of hydrochlorothiazide in tablet dosage forms by using different solvents. Der Pharma Chemica 4(1): 10-14.
- Delhiraj N, Alla B, Azra P, Tahimeen Muzzamil K, Anbazhagan S (2012) Simultaneous spectrophotometric estimation of olmesartan medoxomil, amlodipine besylate and hydrochlorothiazide inpharmaceutical dosage form. Journal of Chemical and Pharmaceutical Sciences 5(1): 30-32.
- Patel R, Patel J (2012) Absorbance ratio spectrophotometric method for the estimation of nebivolol Hcl and hydrochlorthiazide in their combined dosage form. International Journal of Advances in Pharmacy Biology and Chemistry 1(4): 496-502.
- Shailesh G, Arvind U, Sheetal P, Poonam S, Bhavika C, et al. (2014) Development and validation of UV spectrophotometric method for hydrochlorothiazide in bulk drug using mixed hydrotropic solubilization. International Journal of Pharmaceutical Research and Development 6(04): 167-172.
- Idress F, Al-Momani (2011) Determination of hydrochlorothiazide and enalapril maleate in tablet formulations by reversed-phase HPLC. Turkish Journal Chemistry 25: 49-54.
- Zorica V, Nedžad M, Miralem S, Jasmina B, Predrag S (2012) Simultaneous analysis of irbesartan and hydrochlorothiazide an improved HPLC method with the aid of a chemometric protocol. Molecules 17: 3461-3474.
- Dhanalakshmi K, Tatineni V, Nagarjuna R, Jotheiswari D (2013) Analytical method development and validation of telmisartan and hydrochlorothiazide in dissolution by RP-HPLC. International Journal of Biological & Pharmaceutical Research 4(3): 200-211.
- Kaliappan I, Pushpangadhan S (2013) Application of astability HPTLC method for simultaneous quantitative determination of olmesartan medoxomil and hydrochlorothiazide in pharmaceutical dosage forms. Journal of Analytical Methods in Chemistry 2013: 1-8.
- Antil P, Kaushik D, Jain G, Srinivas K, Indu T (2013) UPLC method for simultaneous determination of valsartan & hydrochlorothiazide in drug products. Journal Chromatography Separation Techniques 4(5): 1-5.
- Napa D, Sockalingam A (2014) Validated liquid chromatographic method for the estimation of antihypertensive mixture in pharmaceutical dosage forms. World Journal of Pharmacy and Pharmaceutical Sciences 3(6): 1916-1927.
- Gotardo M, Pezza L, Pezza H (2005) Determination of hydrochlorothiazide in pharmaceutical formulations by diffuse reflectance spectroscopy. Eclética Química 30(2): 17-24.
- Ali F (2014) Electrochemical oxidation behavior of hydrochlorothiazide on aglassy carbon electrode and its voltammetric determination in pharmaceutical formulations and biological fluids. Journal of Food and Drug Analysis 22(3): 363-369.
- Kaya B, Erdal D, Dumitru B (2009) Chemometric methods for the simultaneous spectrophotometric determination of telmisartan and hydrochlorothiazide in the commercial pharmaceuticals. Revista de Chimie 60(6): 544-550.
