 Case Report
Case ReportIntroduction
Down syndrome (DS) and Klinefelter syndrome (KS) are both serious congenital birth defects. DS is estimated to affect 1 in 700 births [1]. KS is one of the most common sex chromosomal abnormalities with the incidence around 1 in 500 to 1 in 1000 male births [2]. The existence of more than one chromosomal abnormality in the same individual could be considered as a rare phenomenon [3]. The first report of the patient with double trisomy combining DS and KS was published by Ford and colleagues in 1959 [4]. The patients with both DS and KS have variable clinical presentations [3]. The main characteristics of DS could be developmental delay, single palmar crease, short stature, facial anomalies, hypotony and short hands. Additionally, cardiac and gastrointestinal defects, hypothyroidism, and celiac disease are commonly associated with DS [3]. Congenital heart defects have found in 40-60% of DS cases [5] but rarely reported in children with KS [6]. This study aimed to report a fetus diagnosed with dual DS-KS who presents the characteristics of congenital heart defect.
Case Report
A 30-year-old woman with normal family history, gravida 2, para 1, underwent a sonographic examination at 29 weeks of pregnancy for the first time at Vinmec Times City International Hospital (Hanoi, Vietnam). Historically, the 12th week ultrasound examination showed no sign of abnormality (NT 2.3mm), and the patient did not take the double test. The triple test showed low risk for chromosomal aneuploidy. At 29th week, the ultrasound analysis revealed the fetal features including complete atrioventricular septal defect (Figure 1) but normal great vessel courses; absent nasal bone (Figure 2); facial profile of DS; bilateral brachy mesophalangia of the fifth digit; a short femur (51mm - 5.6 percentile) and humerus (43mm - <1 percentile) for gestational age (29 weeks and 4 days). The clinical doctor indicated invasive tests including quantitative fluorescence PCR (QF-PCR) and cytogenetic analysis (karyotyping) from amniotic fluid. QF-PCR was performed using Devyser Compact v3 kit (DevyserAB, Sweden). The results were described according to the International System for Human Cytogenetic Nomenclature (ISCN 2016).
For chromosome 21, the QF-PCR result showed the contribution of 3 alleles on the 21A (1: 2 ratio), 21B (1: 1: 1 ratio), 21C (1: 1: 1 ratio), 21D (1: 1: 1 ratio), 21H (1: 2 ratio), 21I (2: 1 ratio) of chromosome 21; normal allele number for chromosome 13 and chromosome 18 based on interpretation of 5 Short Tandem Repeat (STR) markers 13A, 13B, 13K 18B, and 18D. The presence of two X chromosomes and one Y chromosome, rsa (13,18) x2, (21)x3, (X) x2, (Y)x1 corresponding to dual DS-KS. Because congenital heart defect was found in ultrasound, a prenatal BoBs test was also performed to detect other microdeletions related to this symptom and to independently confirm the QF-PCR result. Prental BoBs test showed the presence of three copies of the chromosome 21, two copies of chromosome X and one copy of chromosome Y with no detection of microdeletion. This result is corresponding to dual DSKS. Cytogenetic investigation of the amniotic fluid at 30th week of pregnancy was performed with Amnio max complete media and G-banding of chromosomes. The results were described according to the International System for Human Cytogenetic Nomenclature (ISCN 2016). The cytogenetic analysis revealed a case of double aneuploidy with the karyotype 48, XXY, inv (9) (p11q13), +21 (Figure 3).
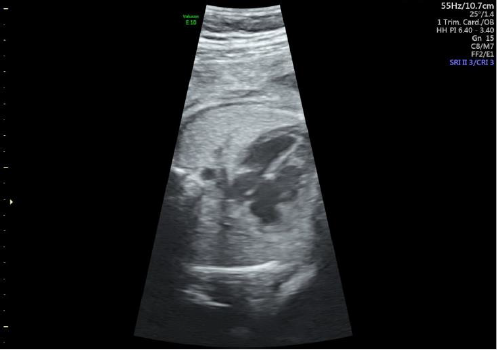
Figure 1: Ultrasound images show complete atrioventricular septal defect.
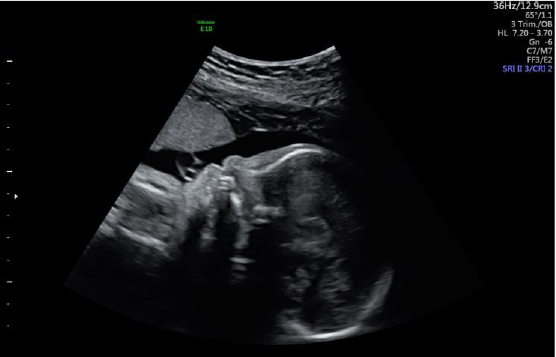
Figure 2: Absent nasal bone.
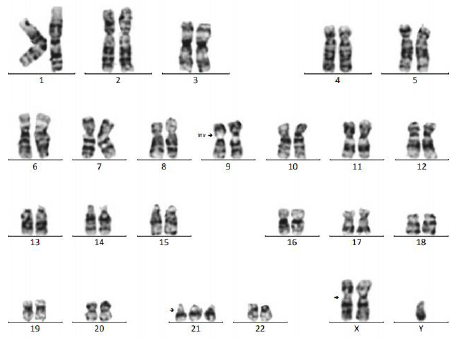
Figure 3: Cytogenetic analysis reveals a double aneuploidy 48, XXY, inv (9) (p11q13), +21.
We provided genetic counseling for the patient and her family about the ultrasound and prenatal tests result as well as the fetus condition. The family decided to terminate the pregnancy at 32nd week of pregnancy. This study was conducted with the approval from the Ethics Committee of Vinmec International Hospital, Vinmec healthcare system, Hanoi, Vietnam. The patient signed an informed written consent before enrollment in the study, according to the ethical guidelines of the Declaration of Helsinki amended in 2008. The first report of the patient with double trisomy combining DS and KS is published in 1959 by Ford and colleagues [4]. To our best knowledge, over hundred cases have been published in the literature, however only about 20 patients have been reported to have prenatal diagnosis. Two chromosomal abnormalities present in the same individual (double aneuploidy) is a rare phenomenon. According to previous studies, the incidence rate of this rare double trisomy is varied from 1 in 94,440 to 350,000 pregnancies [6].
Various methods including screening (ultrasound, biochemical markers, non-invasive prenatal screening), diagnosis (cytogenetic analysis or molecular tests on chorionic villus sampling, amniotic fluid) have been applied to identify the risk of carrying fetus with chromosomal abnormalities for pregnant women [3]. Therefore, early detection will help improve the maternal and fetus health care management. Ultrasound, which can detect both major structural abnormalities and minor “soft markers”, plays a key role in prenatal aneuploidy screening. In fetus affected with DS, sonographic findings can reveal cardiovascular, central nervous, craniofacial, musculoskeletal, gastrointestinal, and urinary tract system anomalies. The major structural abnormalities may include duodenal atresia and cardiac anomalies such as septal defects, tetralogy of Fallot, and atrioventricular canal defects. Soft markers may be seen in normal population but have an increased incidence in fetus with chromosomal abnormalities [3]. Prenatal sonography in the second trimester provides a “genetic sonogram” for detecting structural characteristics of a fetus with DS [3]. The presence of multiple markers may increase the probability of DS. Unfortunately, these anomalies are not always detected by prenatal ultrasound screening.
We reported here a prenatal case with the finding of complete atrioventricular septal defect (AVSD), absent nasal bone, facial profile of DS, bilateral brachy mesophalangia of the fifth digit, a short femur and humerus for gestational age. The positive likelihood ratio of nasal bone absence for DS screening was estimated around 29.00-66.75 [7]. This abnormality is commonly used as soft markers to screen for DS during the first or second trimester ultrasound. Du et al. [7] described 38 cases of DS among the 56 707 singleton pregnancies suggested that the sensitivity and specificity of the absent fetal nasal bone marker in detecting DS were 31.58 and 99.90%, respectively. The positive and negative predictive values were 16.90 and 99.95%, respectively [7]. In the study of Vos, et al. [8], they described two new methods for assessing the relationship between the mandible and maxilla: the maxilla-nasal-mandible (MNM) angle and the fetal profile (FP) line. The MNM angle, defined as the angle between the maxilla-nasal and mandible-nasal lines, is constant throughout pregnancy at about 13.5° in euploid fetuses, whereas the angle was much smaller (8.2°-11.2°) in the fetuses with DS. The fetus in our study has the MNM angle of 11°.
In individuals with double aneuploidy, 48, XXY, +21, the characteristic of DS often predominates and KS phenotype does not occur until puberty [3]. In over 20 prenatal cases in the Table 1, only two of them were found to have the congenital heart defect (CHD). To our best knowledge, only 13 individuals of 48, XXY, +21 karyotype with CHD were reported in the previous studies. Congenital heart defects have found in 40-60% of DS cases including 45% atrioventricular septal defects (AVSD) [5]. Rodrigues et al. [6] reported that the frequency of CHD in dual DS-KS and DS populations seems to be different. The incidence of CHD in DS patients can reach to 60% compare to 15% of that in dual DS-KS patients. Many other syndromes also have CHD including Di George, Williams - Beuren, therefore we also performed Prenatal BoBs test for the amniotic fluid to rule out. Prenatal BoBs test showed negative results for nine tested microdeletion syndromes including Williams - Beuren, Di George - 1 and Di George - 2 syndromes. It is important to analyze a bigger number of patients to establish the correlation between CHD and DS-KS.

Table 1: Prenatal cases of Down-Klinefelter syndrome and the clinical features.
The cytogenetic analysis result from amniotic fluid shows a karyotype of 48, XXY, inv (9) (p11q13), +21, with a normal maternal profile (46, XX) and abnormal karyotype profile from the father (46, XY,1qh+, inv (9) (p11q13)). The pericentric inversion of chromosome 9 is a chromosomal heteromorphic and often found in human. Around 1.6% in the general population has this common chromosome variant [9]. In the current study, the inversion of chromosome 9 was suggested with the association to the pregnancy loss, infertility and/or miscarriage. The incidence of the inversion of chromosome 9 variant was reported to be significantly higher in the infertile population compared with the general population [10]. In our study, no history of infertility and miscarriage (Para 1001) has been recorded in this couple. It is believed that meiotic nondisjunction is the most common reason for the forming of aneuploidy. However, the cause of nondisjunction is unclear. The most favored explanation is the advanced maternal age [3]. In our case, the fetus was delivered by a 30-year-old mother and a 35-year-old father.
Conclusion
In conclusion, prenatal screening and diagnosis plays an important role in providing genetic counseling for the couples having dual DS-KS fetus. It helps in making the decision for pregnancy or improving the prenatal health care management for both mother and baby. Our report contributes to the better understanding of DSKS genotype-phenotype relationship and addresses the importance of extensive methods for diagnosis ranging from conventional ultrasound to cytogenetics and molecular genetic testing.
References
- Mai CT, JL Isenburg, Mark AC, RE Meyer, A Correa, et al. (2019) National population-based estimates for major birth defects, 2010-2014. Birth Defects Research 111(2): 1420-1435.
- Los E, GA Ford (2020) Klinefelter Syndrome, in Stat Pearls. Stat Pearls Publishing LLC.: Treasure Island (FL).
- Aydin C, SE Yalcin, Y Yalcin, HS Selim (2013) An interesting prenatal diagnosis: double aneuploidy. Case reports in obstetrics and gynecology 2013(4): 790286-790286.
- Ford CE (1959) The chromosomes in a patient showing both mongolism and the Klinefelter syndrome. Lancet 1(7075): 709-710.
- Zhang H, L Liu, J Tian (2019) Molecular mechanisms of congenital heart disease in down syndrome. Genes & diseases 6(4): 372-377.
- Rodrigues MA, LF Morgade, LFA Dias, RV Moreira, PD Maia, et al. (2017) Down-Klinefelter syndrome (48, XXY, +21) in a neonate associated with congenital heart disease. Genet Mol Res 16(3).
- Du Y, Y Ren, Y Yan, Li C (2018) Absent fetal nasal bone in the second trimester and risk of abnormal karyotype in a prescreened population of Chinese women. Acta obstetricia et gynecologica Scandinavica 97(2): 180-186.
- Vos FI, EAP De J, M Bakker, E Tromp, KO Kagan, et al. (2015) Fetal facial profile markers of Down syndrome in the second and third trimesters of pregnancy. Ultrasound in Obstetrics & Gynecology 46(2): 168-173.
- Sipek A, Ales P, Romana M, Hrckova L, Suttrova E, et al. (2015) Pericentric Inversion of Human Chromosome 9 Epidemiology Study in Czech Males and Females. Folia Biol (Praha) 61(4): 140-146.
- Bharadwaj M, S Chapadgaonkar (2012) Homozygosity and heterozygosity of pericentric inversion of chromosome 9 and its clinical impact. Journal of Clinical and Diagnostic Research 6.
