 Research Article
Research ArticleABSTRACT
The main task in current drug discovery has been the design and growth of new antibacterial drugs with better efficacy and no side effects. We conducted docking simulation to salicylidene acylhydrazides derivatives as ligands into the crystal structure of IncA as the receptor. The 3D crystal structure of IncA as the receptor was downloaded from PDB (Code ID: 6e7e). The structure of ligands and protein were prepared using Spartan and Discovery studio, respectively. The docking process, the interaction, and binding of ligands – protein was done and visualized using iGemdock software and Discovery studio visualizer. The results showed Van der Waals, Pi-donor hydrogen bond, Pi-pi stacked, and amide-pi stacked interaction between compound 44 (N’‐[(Z)‐(2‐hydroxy‐3,5‐dinitrophenyl)methylidene]‐2‐[(3R,5S,7s)‐adamantan‐1‐yl] acetohydrazid) with Plk1, but this compound have more Van der Waals interaction with His253, His250, Lys246, Arg245, Thr238 and Ser154 of the crystal structure of IncA as the receptor. iGemdock (drug screening) scores of compounds 44, is -87.3827 kcal/mol. It is predicted that compound 44 has potency as a lead compound to find a new anti- Chlamydia trachomatis candidate for possible therapeutic agents.
Keywords: Bacteria; Chlamydia Trachomatis; Docking Simulations; Salicylidene Acylhydrazides Derivatives; Igemdock
Introduction
Chlamydia trachomatis is a typical human microorganism that causes explicitly sent infections, fruitlessness, trachoma, and visual deficiency in individuals everywhere on the world [1]. As indicated by the World Health Organization (WHO), 85 million individuals got anti-microbials for trachoma, a blinding eye contamination that influences 42 nations [2], also, in excess of 100 million instances of explicitly communicated Chlamydia trachomatis are analyzed every year overall [3]. Contaminations with these commit intracellular microorganisms have few therapeutic choices, and the most effective treatment is a single dose of azithromycin [4]. Over time, these drugs lead to side effects such as abdominal pain, diarrhea, and other gastrointestinal disorders in chronic treatment. Tetracycline (e.g., Oxytetracycline and Chlortetracycline) were discovered in the 1940s and exhibited activity against a wide scope of microorganism including gram-positive and gram-negative microscopic organisms, chlamydia, mycoplasma, rickettsia, and protozoan parasites [5]. Like another tetracycline, Chlortetracycline can inhibit bone and tooth mineralization in growing and unborn animals, and color their teeth yellow or brown. It can also unpair liver and kidney function, Allergic reactions are rare [6]. Oxytetracycline can cause gastrointestinal and photosensitive unfavorably susceptible responses. It can likewise harm calcium-rich organs, for example, teeth and bones [7].
Accordingly, exploitation of new potent, safer, and cheaper Chlamydia trachomatis or bacterial inhibitors becomes the biggest challenge the human race is facing nowadays. For this reason, the development of new drugs able to fight Chlamydia trachomatis is still in great interest. Lately, the utilization of molecular modeling has delivered exceptionally amazing outcomes in the medication disclosure measure [8]. To seek after our past work on Chlamydia trachomatis [1] and the molecular modeling study on salicylidene acylhydrazides derivatives [9]. In this place, we report the molecular modeling of salicylidene acylhydrazides derivatives. To pursue, our ongoing research on molecular modeling of Chlamydia trachomatis inhibitors. We aim in this study to find a specific interaction that may provide the best affinity with the protein than the current therapy.
Materials and Methods
In the present study, we have selected 58 salicylidene acylhydrazides derivatives from the literature [9] that have been already validated to have antimicrobial activity. All compounds were tried utilizing a similar test strategy and a wide scope of Chlamydia trachomatis hindrance biological activities were covered. Structures picked out were modify to achieve maximum efficiency in storage capacity, time or cost with the semiempirical PM3 method using the Spartan’14 software (www.wavefun.com) (Table 1). The modify to achieve maximum efficiency in storage capacity (optimized) structures were saved in .pdb format for further studies as docking software takes .pdb format as an input file.
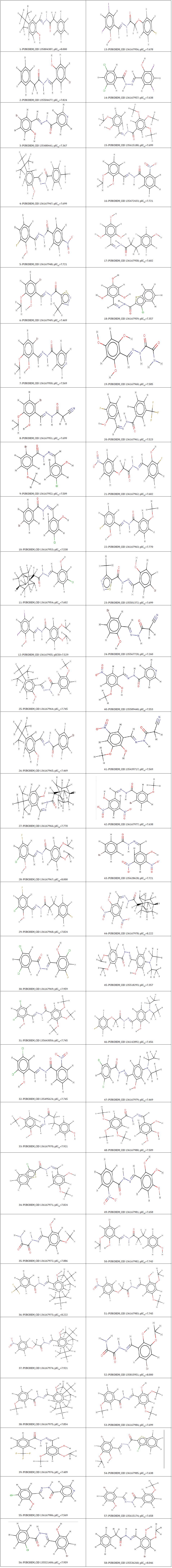
Table 1: PubChem_CID number, corresponding activity pIC50 of salicylidene acylhydrazides derivatives.
Protein Preparation
The strength of association or binding affinity between two molecules can be predicted by molecular docking techniques. In the current study, we have selected 58 phytochemical compounds that have been suggested to possess potential antimicrobial activity. The three-dimensional structure of receptors such as the crystal structure of IncA was retrieved from RCSB (www.rcsb. org/pdb) protein data bank (PDB Code: 6e7e) [10]. The whole structure of the receptors was targeted for our molecular docking study except heteroatoms that were detected from the receptors. Docking analysis was performed using iGemdock, [11] which uses empirical scoring function and Generic Evolutionary Method for molecular docking. It has a graphical user interface that recognizes the pharmacological interactions and performs virtual screening. It has been decided to select the following parameters for docking as Population size: 200, Number of generations: 70, and Number of solutions: 3. The docking simulation interaction was visualized through Discovery studio software.
Results and Discussion
Four anti-chlamydia trachomatis drugs- Ampicillin, Oxytetracycline, Chlortetracycline, and Ceftriaxone (Azithromycin) were used as a control for the following study. The four were docked into the enzyme; iGemdock by calculating their binding energy, Vander Waals energy, electrostatic energy, hydrogen bonding. The post analysis tools of iGemdock works by using K-means and hierarchal clustering methods. (Table 2) shows the summary of docking results. Binding energies of the receptor-ligand interactions are very important to report how fit the drug binds to the target macromolecule. It can be calculated that according to iGemdock docking results Ampicillin is the best binding receptor because of its total energy of -92.5875kcal/mol. The lesser the energy greater will be acceptability of the chemical as a drug. (Figure 1), shows the binding interactions of the controls (drugs) with the studied protein, where Ampicillin (Figure 1a), Oxytetracycline (Figure 1b), and Chlortetracycline (Figure 1c) was found to have unfavorable interactions with the protein. Ceftriaxone (Azithromycin) has a Van der Waals interaction of -55.1418 kcal/mol, electrostatic interaction of -1.65517 kcal/mol, the total binding energy is equal to -82.7819 kcal/mol with no unfavorable bump or donor-donor interaction (Figure 1d).
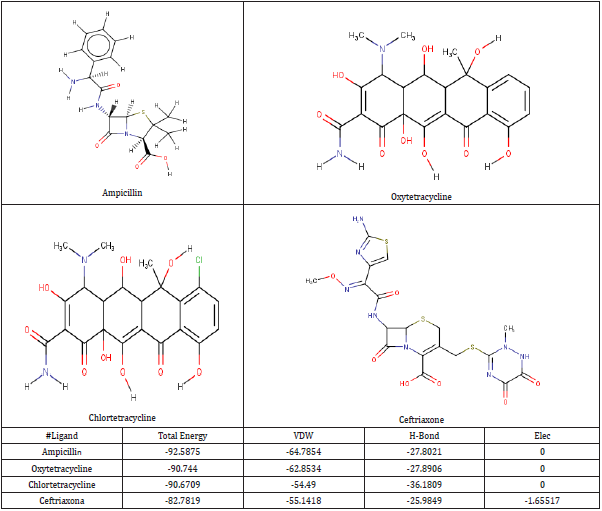
Table 2: Structures and docking results (kcal/mol) of standard in the active site of the protein.
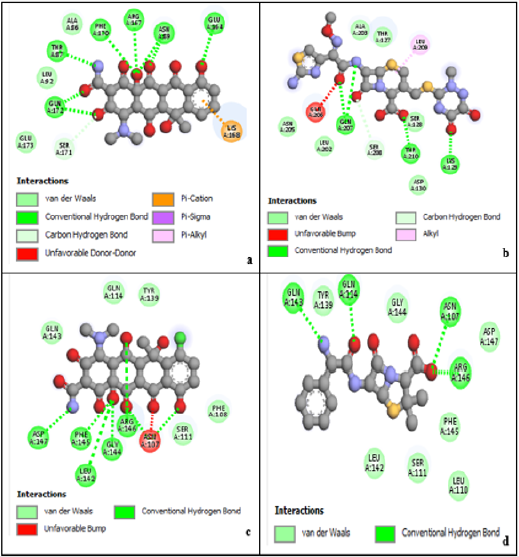
Figure 1: Binding interaction of the control/standard drugs with the macromolecule.
According to research, Ceftriaxone (Azithromycin) has common side effects include pain at the side of injection and allergic reaction. Other possible side effects include gall bladder disease, seizures, hemolytic, anemia, etc. (Table 3) show the molecular docking results of the salicylidene acylhydrazides derivatives were Compound 44 (N’‐[(Z)‐(2‐hydroxy‐3,5‐dinitrophenyl) methylidene]‐2‐[(3R,5S,7s)‐adamantan‐1‐yl]acetohydrazid) has the best binding energy of -87.3827 kcal/mol and (Figure 2) shows the binding interaction with the protein. Compound 44 however, was found to have the lowest Van der Waals interaction ( −87.3827) than the standards (control) and engaged in pi-donor hydrogen bond between HIS249, pi-pi-Stacked and Amide-pi-Stacked interaction. Compound 44 was showing better interaction than the standard or control drugs available in the market. It also showed better binding energy than the Ceftriaxone. The possible binding modes of compound 44 at the target protein active sites have been shown in (Figure 2). The protein residues Ser154, Ile241, Arg245, Thr238, Ala242, His249, Lys246, His250, and His253 were found interacting with the ligand without unfavorable bumps shows that compound 44 is better than the standard drugs with side effects and unfavorable bumps interactions.
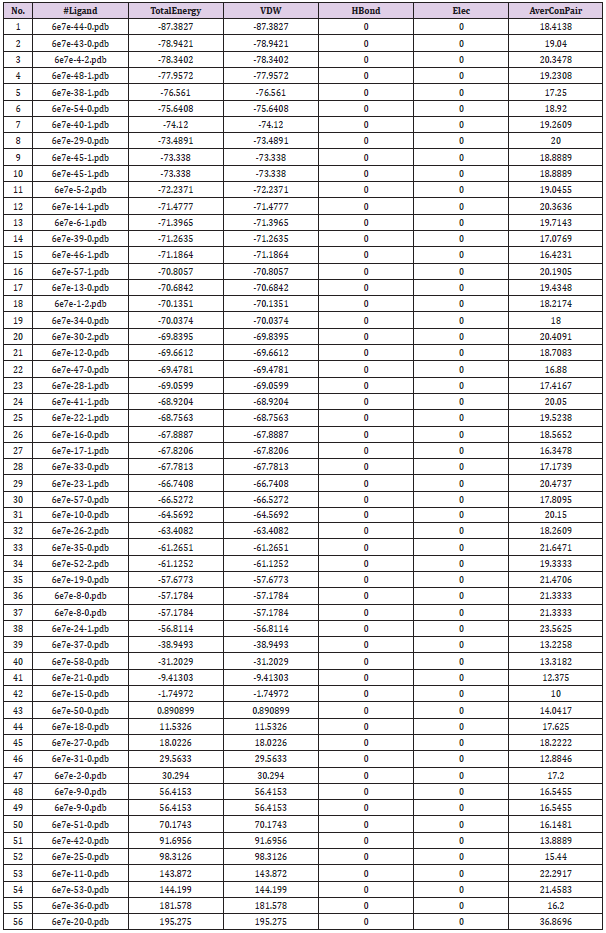
Table 3: Docking results (kcal/mol) of salicylidene acylhydrazides derivatives in the active site of the protein.
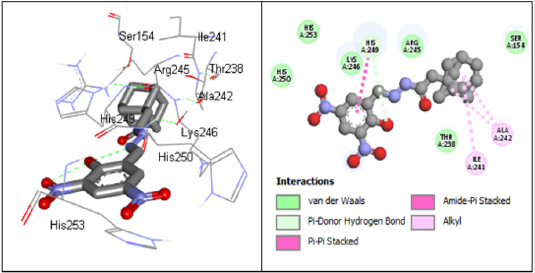
Figure 2: Interaction of compound 44 with the protein structure.
Conclusion
There is an urgent need to explore new therapeutic compounds which will offer safer, efficacious, and cost-effective treatment, that will improve the life quality of patients. In this study, using the molecular docking method, compound 44 (N’‐[(Z)‐(2‐hydroxy‐3,5‐ dinitrophenyl)methylidene]‐2‐[(3R,5S,7s)‐adamantan‐1‐yl] acetohydrazid) has been identified to inhibit Chlamydia trachomatis. Therefore, this compound can be an effective drug candidate for controlling bacteria than the four standard drugs with side effects.
Acknowledgment
The authors gratefully acknowledge the Department of Chemistry, Ahmadu Bello University, Zaria (Samaru, Zaria-Nigeria); for computational studies, and as part of the Ph.D. thesis.
References
- Edache EI, Uzairu A, Mamza PA, Shallangwa GA (2020) Computational Modeling and Molecular dynamics Simulations of Thiazolino 2-pyridone amide analog compounds as Chlamydia trachomatis inhibitor. J Chem Lett 1: 123-138.
- Thabit AA, Al-Khatib T, Hail WHM, Al-Soofi A, Thabit ANA, et al. (2018) Prevalence of trachoma in Yemen: Results of population-based prevalence surveys of 42 evaluation units in nine governorates. Ophthalmic epidemiology 25(1): 62-69.
- Mojica SA, Eriksson AU, Davis RA, Bahnan W, Elofsson M, et al. (2018) Red Fluorescent Chlamydia trachomatis Applied to Live Cell Imaging and Screening for Antibacterial Agents. Frontiers in microbiology 9: 3151.
- Stamm WE (1991) Azithromycin in the treatment of uncomplicated genital chlamydial infections. The American journal of medicine 91(3): S19-S22.
- Chopra I, Roberts M (2001) Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiology and molecular biology reviews 65(2): 232-260.
- Whitaker ED (2017) The Trouble with Human Nature: Health, Conflict, and Difference in Biocultural Perspective. Taylor & Francis.
- Ngangom BL, Tamunjoh SSA, Boyom FF (2019) Antibiotic residues in food animals: Public health concern. Acta Ecologica Sinica 39(5): 411-415.
- Hadni H, Elhallaoui M (2020) 2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents. New J Chem.
- Edache EI, Uzairu A, Mamza PA, Shallangwa GA (2021) Docking Simulations and Virtual Screening to find Novel Ligands for T3S in Yersinia pseudotuberculosis YPIII, A drug target for type III secretion (T3S) in the Gram-negative pathogen Yersinia pseudotuberculosis. Chemical Reviews and Letters 4: 130-144.
- Cingolani G, McCauley M, Lobley A, Bryer AJ, Wesolowski J, et al. (2019) Structural basis for the homotypic fusion of chlamydial inclusions by the SNARE-like protein IncA. Nature Communications 10: 2747.
- Yang JM, Chen CC (2004) Gemdock: A generic evolutionary method for molecular docking. Proteins 55: 288-304.
