 Case Report
Case ReportAbstract
Objectives: Hearing loss is the most common sensory disorder in humans. Approximately 60% of cases of congenital hearing loss have a genetic aetiology. Consequently, the genetic study would significantly improve the diagnostic performance of these hearing impairments avoiding unnecessary tests and helping to establish the evolution, prognosis and treatment of the disease. These studies are of great relevance in cases of syndromic hearing loss.
Methods: Using an own designed panel of next generation sequencing (NGS), we studied the case of a two year old boy, with a congenital bilateral NSHL detected through neonatal hearing screening. The patient did not present any other associated pathology. As the hearing loss was profound and bilateral, it was decided to place a cochlear implant in both ears when he was one year old.
Results: The analysis revealed the presence of the heterozygous mutation c.1066C>T, p. (Arg356*), in the MITF (NM_198159.2) gene which was confirmed by Sanger sequencing. The segregation study of the mutation in the MITF gene was performed in the family and only the father showed the same mutation with a different phenotype.
Conclusion: We describe the clinical spectrum in a family with MITF mutation. They are a clear example of incomplete intrafamilial penetrance and variable expressivity already reported in other cases with MITF mutations.
Keywords: Waardenburg Syndrome; Neurosensorial Hearing Loss; Cochlear Implant; Pigmentary Abnormalities; Retinal Hipopygmentation
Introduction
Hearing loss is the most prevalent sensory impairment in humans, affecting around 5% of the world population [1]. In developed countries it is estimated that more than 60% of cases are due to genetic causes. Therefore, genetic testing has become more important for highly accurate diagnosis, prediction of severity, estimation of associated abnormalities, selection of appropriate habilitation options, prevention, and better genetic counseling.
Hereditary hearing loss can be non-syndromic or syndromic. Non-syndromic hearing loss (NSHL) constitutes 70% of it, currently 134 loci have been determined and over 90 genes causally in it. Syndromic hearing loss (SHL) constitutes 30% of the disease, more than 400 syndromes are known with hearing loss as one of the symptoms and for many of these the causative genes have been identified. Some of the most frequently occurring syndromes involving hearing loss includes Branchio-Oto-Renal syndrome, Waardenburg syndrome, Pendred syndrome and Usher syndromes among others.
Waardenburg syndrome (WS) is an autosomal genetic condition that exhibits a very high penetrance rate, a variable expressivity of clinical features and heterogeneity of loci and alleles. Its main clinical features are congenital hearing loss and pigmentation abnormalities (including depigmented patches of the skin and hair, heterochromia and/or hypopigmentation of the irides). The syndrome is genetically heterogeneous, having been classified into four groups based on clinical grounds [2]. WS type 1 is characterized by eyes that appear widely spaced, congenital hearing loss, and patchy pigment disturbances of the iris, hair and skin. In contrast to WS type 1 (WS1), WS2 patients do not show dystopia cantorum (eyelid anomaly; lateral displacement of the inner canthi of the eye). The phenotype of WS3 and WS4 patients is similar to WS1 but with additional hypoplasia of limb muscles (WS3) or Hirschprung’s disease (WS4) [3].
WS2 is an autosomal dominant syndrome characterized by congenital neurosensorial hearing loss (NSHL), pigmentary abnormalities of the eyes, hair and skin and the absence of dystopia cantorum, the lateral displacement of the ocular inner canthi, which is seen in some other forms of Waardenburg syndrome [4]. WS2 is caused by heterozygous pathogenic variants in genes MITF and SOX10, which together explain nearly 30% of cases [5]. We report the case of a two years old boy with profound NSHL leading to bilateral cochlear implant, which showed a truncating mutation in MITF.
Materials and Methods
Case 1
Patient 1, a boy of two years old, with a congenital bilateral NSHL detected through neonatal hearing screening. The patient did not present any other associated pathology. As the hearing loss was profound and bilateral, it was decided to place a cochlear implant in both ears when he was one year old. Her unaided visual acuity at presentation was 20/41 Binocular (Teller test). Her ocular examination did not show dystopia cantorum (W index -0.27 mm) or heterochromia iridis. He did not present nystagmus. While both eyes anterior segment were normal, both fundoscopies were albinotic, showing retinal hypopigmentation. Refraction under cycloplegic drops was within normal limits. Therefore, the patient also had no other pigmentary abnormalities of the eyes, hair and skin.
In order to make a diagnosis, it was decided to carry out a genetic study of the patient. Written informed consent was obtained from the index case and other family members. Molecular study was performed, after DNA extraction, through next generation sequencing (NGS) using the Trusight One panel (Illumina), targeting disease-associated regions of the exome. The analysis revealed the presence of the heterozygous mutation c.1066C>T, p. (Arg356*), in the MITF (NM_198159.2) gene which was confirmed by Sanger sequencing (Figure 1).

Figure 1: Chromatograms of part of MITF gene sequence showing the presence in heterozygosis (A) and the absence (B) of the c.10661C>G, p.(Arg356*) mutation in individuals III:8 and II:7 respectively.
Case 2
Patient 2, father of the index case, reported the presence of unilateral hearing loss in the left ear. To evaluate the hearing deficit, an auditory brainstem response and an audiometry was done, which showed a left profound unilateral sensorineural hearing loss. The hearing in the right ear turned out to be normal. An imaging test was carried out with a magnetic resonance imaging, finding no alterations related to the disease. Ophthalmological history included pars plana vitrectomy OD in the context of rhegmatogenous retinal detachment and cataract surgery.
Her aided visual acuity at presentation was 20/40 OD and 20/40 OS. On ocular examination, neither dystopia cantorum (W index -0.107 mm) nor iris pigmentary abnormality were noted. OD had pseudophakia and OS anterior segment was normal. Right fundus showed laser scars at the temporal quadrant whereas left fundus showed hypopigmentation only in periphery and both of them myopic choroidosis. Axial length was 21.9mm both eyes.
Segregation study of the mutation in the MITF gene was performed after DNA extraction through Sanger sequencing showing the presence of the mutation c.1066C>T, p. (Arg356*), in heterozygotic state.
Other Family Cases
The other paternal family members were examined by expert clinicians in audiological (tonal and verbal audiometry, Brain stem evoked potentials and otoacoustic emissions), ophthalmological (visual assessment, presence of heterochromia and retinal fundus and macula examination), neurological and dermatological terms.
Hearing tests were normal in all of them, except for a case of presbycusis in a paternal uncle over 50 years of age. Dystopia cantorum was absent in all affected individuals, with a W index <1.90. In the rest of the family, only a paternal uncle presented pigmentary abnormalities of the hair. The genetic study for the c.1066C>T, p. (Arg356*) in the MITF gene was negative in all living relatives (Figure 2). It should be noted that the genetic study could not be carried out on the paternal grandparents of the index case, because they had died a few years before. Therefore, it was not possible to know if the mutation was inherited or de novo.
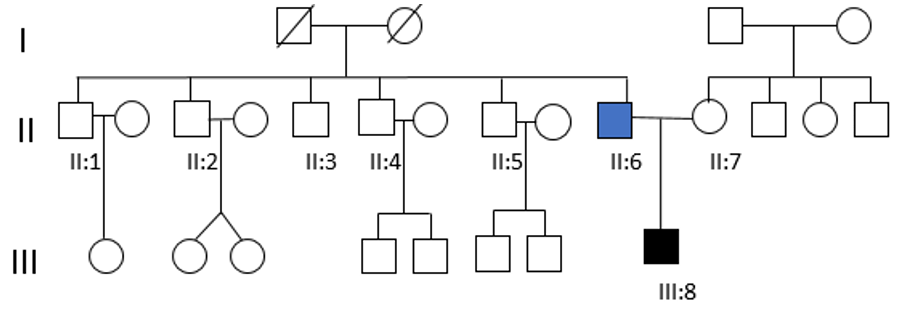
Figure 2: Pedigree of the Waardenburg syndrome family showing congenital bilateral NSHL (III:8) and unilateral hearing loss (II:6).Both of them carry the mutation c.1021C>G in MITF gene. None of the other tested family members (II:1, II:2, II:3, II:4, II:5 and II:7) showed the presence of the nonsense mutation.
Discussion
Hearing loss (HL) is one of the most common sensory defects birth in developed countries and is clinically and genetically heterogeneous. There are at least 30 autosomal loci for nonsyndromic hearing impairment and more than 400 disorders in which hearing impairment is a characteristic of the syndrome. Waardenburg syndrome (WS) is a clinically and genetically heterogeneous disease accounting for >2% of the congenitally deaf population. It is characterized by deafness in association with pigmentary anomalies and various defects of neural crestderived tissues, including hair and eyes. Ocular symptoms include iridial heterochromia, hypopigmentation of the fundi [6-8] and in rare cases unilateral macular degeneration have also been observed8.The pigmentation observed by fundus is dependent on the presence of melanin in both retinal pigment epithelium (RPE) cells and choroidal melanocytes, both of which are affected by MITF mutations.
We have found mutation in the MITF gene in a patient and his father who presented hearing loss as the only symptom. MITF transactivates the gene for tyrosinase, a key enzyme for melanogenesis, and is critically involved in melanocyte differentiation. Absence of melanocytes affects pigmentation in the eyes, skin and hair, and the function of the inner ear. Pathogenic variants in MITF are also reported to cause only NSHL, in particular the mutation in c.1066C>T, p. (Arg356*), in heterozygotic state. A recent study in pigs suggests that MITF gene mutation only affects hair cells of the saccule but has no effect on other vestibular organs. The study also indicated that the survival of cochlear and saccular hair cells was dependent on the potassium release from the cochlear vascular stria, but hair cells of the utricle and semicircular canals were independent on stria vascular [9]. Our patients had hearing loss typical of the cochlear impairment described in WS2. There were no associated vestibular disorders.
Hearing loss and hypopigmentation and in Waardenburg syndrome are thought to be the results of an abnormality of melanocyte differentiation caused by MITF mutations [10]. Our index patient had retinal hypopigmentation in addition to hearing loss. The father did not present any alterations due to hypopigmentation. García-Llorca et al. [11] described in mice how MITF mutations affect both types of pigment cells in the eye: neural crest-derived melanocytes and neuroepithelium-derived RPE cells [11-14]. MITF promotes RPE differentiation, regulates the proliferation of the RPE during development and consequently their retinal function [14]. This study provides more evidence that a functional RPE is important for normal photoreceptor function. Thus, mutations in genes expressed in the RPE such as MITF and OTX2, can have profound effects on retinal structure and function, as a consequence of alterations in the RPE [15].
In humans, MITF mutations have been found in patients with the pigmentation and deafness disorders Waardenburg (OMIM # 193510)5. Hypopigmentation observed in the fundus is not necessarily an indication of RPE dysfunction or retinal degeneration. However, it is of course possible that despite lack of morphological changes, these mutations affect the expression of important downstream target genes affecting photoreceptor function or integrity at older ages. Our index patient had retinal hypopigmentation in both eyes but no visual disturbances at the time of the examination. It will be necessary to perform visual controls in the future to see the clinical evolution of the described finding. The father did not present any ocular alteration associated with WS2.
We have not found any relationship in the literature between the heterozygous mutation c.1066C>T, p. (Arg356*) in MITF and the phenotype observed in our index (bilateral profound sensorineural hearing loss and retinal hypopigmentation). This mutation would also explain the phenotype observed in the father. Thus, we endorse the great heterogeneity that this syndrome may present. WS is characterized by significant intra- and inter-familial phenotypic variability and incomplete penetrance. Somashekar, et al. [16] describes c.1066 C > T mutation in heterozygous state, present in our patient in a family whit a proband with unilateral hearing loss, another proband with bilateral hearing loss and also in other unaffected members. We describe a family affected of the same pathogenic nonsense variant. We have found differences in the phenotype of patients. The index case (1) is affected of bilateral hearing loss and retinal hypopigmentation. The case 2, father of the index case, harbored the same variant in heterozygous state but reported unilateral hearing loss without hypopigmentation abnormalities.
A three-generation family from North of Iran with the same mutation c.1066 C > T, with 9 people with WS2 and 4 normal carriers was reported [17]. We have not found any genetic alterations in all living relatives, but the genetic study was not possible in the paternal grandparents because they were dead at the time of the study, so it could not be ruled out that the mutation was de novo or inherited. In summary, this family is a clear example of incomplete intrafamilial penetrance and variable expressivity already reported in other cases with MITF mutations. We would like to highlight the importance of analyzing this gene in patients who only present with hearing loss.
References
- (2011) World Health Organization. Millions of people in the world have hearing loss that can be treated orprevented.
- Pretell Bocángel M, Souto Melo U, Ucela Alves L, Leandro Ucela Alves, Eliete Pardono, et al. (2018) Waardenburg syndrome: Novel mutations in a large Brazilian sample. European Journal of Medical Genetics 61(6): 348-354.
- Grill C, Bergsteinsdóttir K, Ögmundsdóttir MH, Pogenberg V, Schepsky A, et al. (2013) MITF mutations associated with pigment deficiency syndromes and melanoma have different effects on protein function. Hum Mol Genet 22(21): 4357-4367.
- Waardenburg PJ (1951) A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet 3(3): 195-253.
- Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, et al. (2010) Review and update of mutations causing Waardenburg syndrome. Hum Mutat 31(4): 391-406.
- Bansal Y, Jain P, Goyal G, Singh M, Mishra C (2013) Waardenburg syndrome–a case report. Cont Lens Anterior Eye 36(1): 49-51.
- Mullner Eidenbock A, Moser E, Frisch H, Read AP (2001) Waardenburg syndrome type 2 in a Turkish family: implications for the importance of the pattern of fundus pigmentation. British Journal of Ophthalmology 85: 1384-1386.
- Shields CL, Nickerson SJ, Al Dahmash S, Shields JA (2013) Waardenburg syndrome: iris and choroidal hypopigmentation: findings on anterior and posterior segment imaging. JAMA Ophthalmol 131(9): 1167-1173.
- Du Yi, Li Li Ren, Qing Qing Jiang, Xing Jian Liu, Fei Ji, et al. (2019) Degeneration of Saccular Hair Cells Caused by MITF Gene Mutation. Neural Development 14(1): 1.
- Tachibana M, K Takeda, Y Nobukuni, K Urabe, JE Long, KA Meyers, et al. (1996) Ectopic Expression of MITF, a Gene for Waardenburg Syndrome Type 2, Converts Fibroblasts to Cells with Melanocyte Characteristics. Nature Genetics 14(1): 50-54.
- García Llorca A, Aspelund SG, Ogmundsdottir MH, Steingrimsson E, Eysteinsson T (2019) The microphthalmia-associated transcription factor (Mitf) gene and its role in regulating eye function. Sci Rep 9(1):
- Bharti K, Gasper M, Ou J, Brucato M, Gronenborn KC, et al. (2012) A regulatory loop involving PAX6, MITF, and WNT signaling controls retinal pigment epithelium development. PLoS Genet 8: e1002757.
- Moore KJ (1995) Insight into the Microphthalmia Gene. Trends Genet 11(11): 442-448.
- Nakayama A (1998) Mutations in microphthalmia, the mouse homolog of the human deafness gene MITF, affect neuroepithelial and neural crest-derived melanocytes differently. Mech Develop 70(1-2): 155-166.
- Wen B, Li S, Li H, Chen Y, Ma X, et al. (2016) Microphthalmia-associated transcription factor regulates the visual cycle genes Rlbp1 and Rdh5 in the retinal pigment epithelium. Sci Rep 6: 21208.
- Somashekar Puneeth H, Katta M Girisha, Sheela Nampoothiri, Kalpana Gowrishankar, Radha R Devi, et al. (2019) Locus and Allelic Heterogeneity and Phenotypic Variability in Waardenburg Syndrome. Clinical Genetics 95(3): 398-402.
- Alehabib, Elham, Somayeh Alinaghi, Fatemeh Pourfatemi, Hossein Darvish (2020) Incomplete Penetrance of MITF Gene c.943C>T Mutation in an Extended Family with Waardenburg Syndrome Type II. International Journal of Pediatric Otorhinolaryngology 135: 110014.
