 Research Article
Research ArticleAbstract
Introduction: Cognitive dysfunction is just one of the detrimental consequences associated with ketamine abuse. Increasing evidence indicates that ketamine abuse induces tau hyperphosphorylation and thus leads to memory impairment. In this study, we explored the potential damage to the brain caused by recreational ketamine abuse in comparison with Alzheimer’s disease (AD).
Methods: Here, we investigated the effects of different length exposure of ketamine with and without ethanol on mRNA expressions of the tau hyperphosphorylation and β-amyloid (Aβ) deposition related genes in the rat’s hippocampus and striatum. We also examined the effects of the combined drug treatment on the phosphorylating tau protein at multiple sites and Aβ accumulation, as well as related protein levels in the same brain areas of the rats.
Results: We found significant increases in tau hyperphosphorylation, mediated by the GSK-3β signaling pathway, at S396 and S404 in the hippocampus of rats treated with ketamine and ethanol 6 months. Co-administration of Ketamine and ethanol for 3 months significantly promoted the accumulation of Aβ in the striatum of rats, showing an increase of 91.4% compared to controls. This finding was accompanied by elevated β-site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1) mRNA transcription. Furthermore, repeated ketamine treatments and co-treatment of ketamine with ethanol induced compensatory up-regulation of N-methyl-D-aspartate receptors (NMDAR) expression.
Discussion: The results indicate that recreational ketamine abuse with and without ethanol both lead to neurotoxicity analogous to Alzheimer’s disease. Therapy for damage to the brain induced by ketamine abuse might draw from the gained in AD investigations.
Keywords: Ketamine Recreational Abuse; Cognitive Impairment; Tau Hyperphosphorylation; Aβ; Alzheimer’s Disease
Abbreviations: AD: Alzheimer’s Disease; APP: Amyloid Precursor Protein; NMDAR: N-Methyl-D-Aspartate Receptors; MAPK: Mitogen-Activated Protein Kinase; SD: Sprague Dawley
Introduction
Derived from phencyclidine (piperidine, PCP), ketamine acts as a noncompetitive blocker of N-methyl-D-aspartate (NMDA). The psychological effects of ketamine have been defined as relating to sensory deprivation, mood elevation, and cognitive impairments [1]. Ketamine has been applied in medical use and psychological research for alleviating adverse symptoms stemming from the pathophysiology of depression. On the other hand, it has become a street drug used recreationally in many countries, raising concerns of public health officials worldwide [2]. The prevalence of ketamine use was found to be 1.5% among 12th-grade students in the US; in mainland China, the use of synthetic drugs has increased from 5.6% to 53.8% (2003 to 2010) [3]. Upon three consecutive days of the administration, recreational ketamine users display semantic memory impairment and dissociative and schizotypal symptomatology, associated with ketamine’s action through NMDA receptors [4,5]. Studies have also shown that ketamine’s use at an anesthetic or sub-anesthetic dose could impair the cognitive processes involved in learning and memory, suggesting that nonassociative taste memories may be disrupted by NMDA receptor blockade [6,7]. Current animal research indicates that repeated ketamine exposure augmented NMDA receptor subunit gene expression, notably subunit 1 (NMDAR1 or GluN1). Full sequencing of NMDA receptor genes may help to understand the individual’s vulnerability to ketamine abuse [8].
NMDA receptors, which are vital for learning and memory, are known to be involved in neurotoxic events. NMDA receptors are also hypothesized to play instrumental roles in the pathophysiology of Alzheimer’s disease (AD) [9]. In the prefrontal and entorhinal cortical sections of mouse and monkey brains, significant increases in hyperphosphorylated tau protein were observed after 6 months of ketamine administration. [10] Several kinases and phosphatases have been reported as contributing to tau hyperphosphorylation, including the kinases glycogen synthase kinase-3 (GSK-3) and cyclin-dependent kinase 5 (CDK-5). In contrast, protein phosphatase 2A (PP2A) appears to be the most plausible phosphatase involved in the abnormal posttranslational modification – phosphorylation and methylation [11,12]. Besides, ketamine reduced β-amyloid protein (Aβ) degradation via suppressing neprilysin expression through the dephosphorylating of p38 mitogen-activated protein kinase (MAPK) mediated pathway, which is thought to promote Aβ deposition in patients with AD [13]. There are two pathophysiological hallmarks of AD: the deposition of intracellular neurofibrillary tangles that are aggregates of hyperphosphorylated tau and extracellular plaques that are deposits of amyloid-beta (Aβ) [14]. The question is whether the cognitive impairment of ketamine abuse is related to similar biological events relevant to AD pathogenesis.
Our group recently found a significant increase in dopamine (DA) concentration of rats’ striatum accounted for drug-paired place preference induced by ketamine and ethanol co-administration [15]. Aβ stimulates dopamine release from dopaminergic axons in the anterior cingulate cortex, and excessive dopamine over activates D1 receptors in fast-spiking interneurons, thus contributing to gamma-aminobutyric acid (GABA) inhibitory and excitation/ inhibition imbalance caused by Aβ [16]. These exciting results bring up several points of investigation:
(a) Whether the aggregation of hyperphosphorylated tau in the rat brains is mediated through the GSK-3 pathway; [17]
(b) If the deposition of Aβ in the specific brain area of rats is associated with the activity of β-site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1); [18]
(c) The degree to which the NMDAR subunit expression is modulated in the rat central nervous system after recreational ketamine abuse.
As ketamine and ethanol are recreationally consumed together, we focused on the synergic effects of co-administration of ketamine with ethanol on the rat striatum and hippocampus. In this study, we measured the expression profile of the tau hyperphosphorylation and Aβ deposition-related genes (GSK-3β, Protein phosphatase 2 (PP2A) and BACE1), the levels of tau phosphorylation at serine residues 396/ 404 (Ser 396/404) and threonine residues 231 (Thr 231), and the phosphorylation of GSK-3β. We also examined the expression of NMDAR subunits (NMDAR1 or GluN1, NMDAR2B or GluN2B), Aβ expression, and total tau protein. To our knowledge, this study is the first to scrutinize the cognitive impairment induced by the co-administration of ketamine and ethanol from an angle of AD-related biological events.
Materials And Methods
Animals
Mature male Sprague Dawley (SD) rats (250 g, Laboratory Animal Centre, KMU, People’s Republic of China) were housed at 22°C-24°C with 45%-55% humidity and a 12-hour alternating lightdark cycle with access to food and water ad-libitum. All animal experiments were carried out according to laboratory animal care principles established under China’s law and were also approved by the Ethics Committee of KMU.
Experimental Design
Ninety-six Rats were randomly divided into two main groups: administering drugs daily for 3 months and 6 months. Each main group of rats was divided into four subgroups (two experimental groups and two control groups, n = 12 rats in total). Ketamine (Cat# H35020148, Ketamine Hydrochloride Injec¬tion, Fujian Gutian Pharmaceutical Co, Fujian, People’s Republic of China) was administered intraperitoneally once per day for two experimental groups at the sub-anesthetic dose of 30 mg/kg, according to the suggested dosage: [19,20] ketamine only (K30) and administration of ketamine intraperitoneally at the dose of 30 mg/kg with 20% ethanol (Cat# GB/678-2002, Changshu Yangyuan Chemical Industry Co., Ltd, Changshu, People’s Republic of China, analytical reagent) immediately at a dose of 0.3156 g/kg once a day (K30+E). The two control groups (Control and Ethanol) received the same dose of ethanol and saline. The experimental conditions of each group can be seen in (Table 1). The body weights of the rats were measured weekly, and doses were adjusted appropriately [12]. rats in each group were decapitated under deep anesthesia using diethyl ether at the end of 3 months and 6 months. The hippocampus and striatum were dissected on ice and chosen randomly for evaluating the transcription levels of the genes (n = 4), expression of proteins (n = 4), and density of positive cells (n = 4).
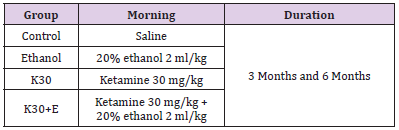
Table 1: Animal models.
Quantitative Real-Time Polymerase Chain Reaction
qRT-PCR was performed as previously reported [15]. Each sample was analyzed in triplicate. Total RNA from the hippocampus and striatum of the SD rats were extracted with TRIzol reagent according to the manufacturer’s instructions (Cat#15596-026, Invitrogen, USA). The RNA concentration and purity (OD260/280) were determined using Nanodrop ND1000 Spectrophotometer (Nanodrop Technologies, Inc. USA). First-strand cDNA was synthesized by using All-in-OneTM miRNA First-Strand cDNA Synthesis Kit (Cat# AMRT-0020, GeneCopoeia, USA) in a 20-μL reaction volume with 1 μg RNA template. A 25-μL total PCR reaction mix contained 12.5 μL All-in-OneTM miRNA qPCR Mix (Cat# AMRT- 0020, GeneCopoeia, USA), 1 μL cDNA, 0.75 μL Forward Primer (10 μM), 0.75 μL Reverse Primer (10 μM), and 10 μL nucleasefree water. We designed the PCR primer sequences using Primer Premier 5.0 (Premier Biosoft International, USA), as shown in (Table 2). The amplification reactions were completed in 96 well plates using the 7900HT Fast Real-Time PCR System (Cat# 4346906, Applied Biosystems, USA). The thermocycling conditions were as follows: denaturation at 95 °C for 10 min, 40 cycles of denaturation at 95 °C for 10 s, and annealing/extension at 72 °C for 30 s. The specificity of amplification was characterized using a melting curve analysis. Differential gene expressions between the drug and saline groups were calculated using the 2-△△CT method with GAPDH as an endogenous control. Relative quantification of GSK-3β, PP2A, and BACE1 mRNA expression was achieved as previously described [21].
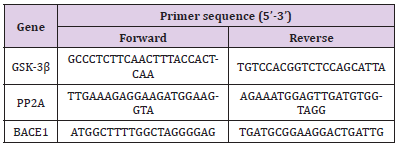
Table 2: Primers for quantitative real-time PCR analysis.
Western Blot Analysis
Total proteins in the interested tissue were extracted with RIPA lysis buffer containing protease and phosphatase inhibitors cocktails (Cat# 58725, Cell Signaling Technology, Inc., USA) and homogenized. Lysates were centrifuged at 12,500 rpm for 15 minutes at 4°C, and the supernatant containing protein extracts was collected. Protein concentrations were determined with the bicinchoninic acid (BCA) Protein Assay Kit (Cat# BCA-1, Sigma- Aldrich, USA). Samples were loaded into each well of a 10% SDSPAGE gel along with 4 μL of Precision plus protein kaleidoscope prestained protein standards (Cat #1610375, Bio-Rad Laboratories, Inc. USA). Proteins were separated, then transferred to a 0.45 PVDF membrane at 25V for 40 minutes. After blocking with 5% non-fat milk in Tris-buffered saline-Tween-20 at room temperature for 2 h, the membranes were incubated overnight at 4°C with following primary detection antibodies separately: β-actin (1:3000, Cat# MA1115, Bosterbio, USA), p-Tau (Thr231) (1:3000, Cat# GTX50174, GeneTex, Inc., USA), p-Tau (Ser396) (1:5000, Cat# EPR2731, Abcam, USA), p-Tau (Ser404) (1:3000, Cat# GTX50176, GeneTex, Inc., USA), GSK-3β (1:3000, Cat# PK1111, Millipore, USA), p-GSK-3β (Ser9) (1:3000, Cat# GTX86837, GeneTex, Inc., USA), and PP2A (1:3000, Cat# 07-250, Millipore, USA). After washing, the membranes were incubated with appropriate horseradish peroxidase-conjugated secondary detection antibodies (Goat anti-Rabbit lgG, Peroxidase Conjugated, 1:5000, Cat# AP132P, Millipore, USA or Goat anti-Mouse lgG, Peroxidase Conjugated, H+L, 1:5000, Cat# AP124P, Millipore, USA) at room temperature for 1 h. The blots were visualized using a Bio-Rad ChemiDoc Touch. Quantitative analysis was operated by Image Laboratory version 6.0 and Image J 1.8.0.
Immunohistochemistry
Rats were anesthetized with diethyl ether and sacrificed by the chest opening, followed by perfusion of 4% paraformaldehyde in phosphate-buffered saline (PBS). The striatum and hippocampus of rats were dissected out and immersed in 4% paraformaldehyde in PBS for further fixation at 4°C overnight, then were embedded in paraffin wax, and coronal sections (5 μm) were cut. Paraffin sections were de-waxed and rehydrated before antigen retrieval with 0.01M citrate buffer in a water bath (60°C). The slides were treated with 0.3% hydrogen peroxide (H2O2) in methanol for 10 minutes to inactivate endogenous peroxidases. After blocking with 1% BSA and 10% normal rabbit serum at room temperature for 1 hour, the slides were incubated with the following primary detection antibodies separately: Anti-Tau (1:800, Cat# ab32057, Abcam, USA), Anti-beta-Amyloid (1:150, Cat# NM26-2.1.3, AnaSpec, USA), Anti-NMDAR1 (1:200, Cat# MAB363, Millipore, USA), Anti- NMDAR2B (1:100, Cat# ab216621, Abcam, USA) overnight at 4°C. Next, slides were incubated with solvent 1 (Polymer Helper, Cat# PV900, ZSGB-BIO) at room tempera¬ture for 20 minutes and then incubated with solvent 2 (polyperoxi¬dase-anti-mouse/rabbit IgG, Cat# PV900, ZSGB-BIO, Beijing, People’s Republic of China) diluted 1:1,000 in 1% BSA at room temperature for 30 minutes. All slides were washed twice (2 minutes per wash) with PBS after incubation with H2O2, primary antibody, solvent 1, and solvent 2. For visualization, the tissue sections were treated with a DAB kit (Cat# Zli-9018, ZSGB-BIO) at room temperature for 3 minutes, and the reactions were quenched with running tap water. After staining, the sections were dehydrated through an alcohol series, cleared with xylene, and mounted. The positive cells were observed using a confocal imaging system with 200× magnification (Olympus FV- 1000; Olympus). The images were quantified using Imagine-Pro Plus 6.0 software analysis system (Media Cybernetics, Rockville, MD, USA).
Statistical Analyses
All data are expressed as the means ± SEM. All statistical analysis was performed using GraphPad Prism 6.0 (GraphPad Software Inc., USA) for Windows. Subsequent analyses involved repeated measures analysis of variance (rmANOVA) in confirming the interactions between the length of exposure and different treatments. When significant, analyses of variance were followed by group comparisons using Tukey’s post-hoc-tests. p < 0.05 was considered statistically significant.
Results
As recapitulated in (Supplementary Table 1), we briefly identified the interactions between two lengths of exposure and four different treatments in rats’ hippocampus and striatum. We observed that interactions between drug administration lengths and drug treatments significantly affected genes and protein expression in the interested brain areas of rat models assessed using qRT-PCR, western-blot, and immunohistochemistry. Different ketamine treatments altered the mrna levels of the tau hyperphosphorylation and aβ deposition related genes. We explored the time-dependent effects of ketamine with and without ethanol on the mRNA expression levels of the tau hyperphosphorylation related genes (GSK3β and PP2A) and Aβ deposition related gene (BACE1) in the hippocampus and striatum of the rats using qRT-PCR. As shown in (Figure 1), compared with the control group, treatment with ketamine (30 mg/kg; 3 months) significantly increased the levels of all three mRNA in rat hippocampus, increasing GSK3β mRNA by 48.12% (Figure 1A), p = 0.026 versus K30+E; 3 months), PP2A mRNA by 64.43% (Figure 1B), p = 0.0008 versus control, p = 0.0013 versus K30+E; 3 months), and BACE1 mRNA by 23.81% (Figure 1C), p = 0.0005 versus K30; 6 months). After exposure to drugs for 6 months, compared with the control group, treatment with ketamine (30 mg/kg; 6 months) decreased the levels of all three mRNA in rat hippocampus without statistical significance.
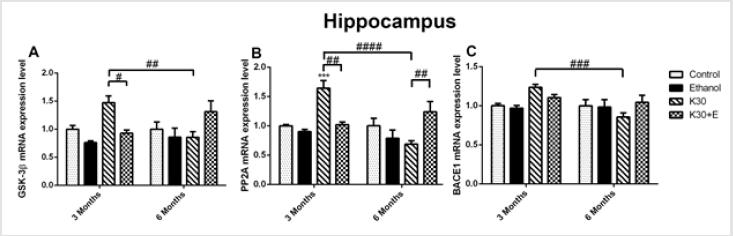
Figure 1: The effects of different ketamine treatments (with and without ethanol) on the mRNA levels the tau hyperphosphorylation and Aβ deposition related genes in the hippocampus of rats (n = 4, per group). All data are presented as mean ± SEM. (**p < 0.01 versus controls and # p < 0.05, ## p < 0.01, #### p < 0.0001 versus drugs administration group).
However, an intriguing and significant upregulation of all mRNA expressions in rat hippocampus were observed in the K30+E group. Measured up to the control, co-administration of ketamine with ethanol elevated GSK3β mRNA by 35.15% (Figure 1A) and PP2A mRNA by 26.41% ((Figure 1B), p = 0.0073 versus K30; 6 months). BACE1 expression was not altered, except for a decrease in the K30 group by 14.90% without statistical significance (Figure 1C). Significant time-dependent decrease of GSK3β, PP2A, and BACE1 mRNA expression was only found for the K group. In the striatum of 3-month groups, as shown in (Figure 2), compared with the control, we found an increase in GSK3β mRNA expression in the K30 group ((Figure 2A), 53.04%, p = 0.0007 versus K30; 6 months). A significantly elevated level of PP2A mRNA was observed in the K30+E group ((Figure 2B), 83.46%, p = 0.0083 versus control). Additionally, a critical increase in BACE1 mRNA expression was noticed in the K30+E group ((Figure 2C), 61.20%, p = 0.0212 versus control). The alterations demonstrate the selectivity of the response to different ketamine treatment conditions. In contrast to the 3 months of drug administration, all genes mRNA levels were unaltered or decreased by different ketamine treatments for 6 months without statistical significance. Time-dependent decreases of GSK3β, PP2A, and BACE1 mRNA expressions were found for the K and K+E groups. Different ketamine treatments increased tau protein hyperphosphorylation regulated by GSK-3β signaling pathway
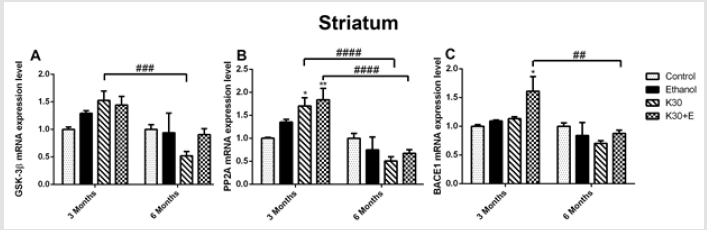
Figure 2: The effects of different ketamine treatments (with and without ethanol) on the mRNA levels the tau hyperphosphorylation and Aβ deposition related genes in the striatum of rats (n = 4, per group). All data are presented as mean ± SEM. (*p < 0.05 versus controls and ## p < 0.01, ### p < 0.001, versus drugs administration group).
A published study has indicated that ketamine induces an increase of tau protein phosphorylation at serine 404 in the hippocampus of neonatal rats. [22] In the present study, we extracted the total protein of rat brains in the interest of exploring hyperphosphorylated tau (Thr231, Ser396, and Ser404) and related proteins involved in the GSK-3β signaling pathway through Western blot, and representative western blot images were shown in (Figure 3) and (Figure 4). After co-administration of drugs for 3 months, as shown in (Figure 3A), only the level of p-tau (Thr231) was significantly higher in the hippocampus of the K30+E group compared to the control group and two ketamine administration groups (increased by 70% compared with controls, p = 0.0002 vs. control). A significant increase in expression of p-tau (Ser396) was observed in the K30 group (increased by 90% compared with controls, p = 0.0074 versus control). However, this effect decreased in the same group after 6 months (Figure 3B). The highest increased expression of p-tau (Ser404) was found in the K30+E group after 6 months (increased by 88.8% compared with control, p = 0.0486 versus controls, (Figure 3C). As GSK-3β is involved in the hyperphosphorylation of tau 17, we investigated the expression of GSK-3β, p-GSK-3β, and PP2A protein. Similar changes with the expression of p-tau (Ser404), the considerably increased expression of GSK-3β was observed in the K30+E group with drug treatment for 6 months (increased by 75.1% compared with control, p = 0.0104 versus controls, (Figure 3D)).
On the contrary, we observed the opposite alteration of p-GSK- 3β protein levels in each drug administration group corresponding to the variation of GSK-3β protein but without statistical significance compared with controls (Figure 3E). There was no visible alteration of PP2A protein expression observed in all drug treatment groups (Figure 3F). Western blot analysis showed that similar changes of p-Tau (Thr231) levels in the striatum across different groups were observed after 3 and 6 months of drug administration. However, the results did not reach statistical significance (Figure 4A). Surprisingly, compared with controls, the highest level of tau phosphorylation at serine 396 was observed in the E group after 3 months of drug administration (increased by 153.4%, p = 0.0001). As well, ketamine with or without ethanol induced the elevated protein levels (K group: increased by 127%, p = 0.0013; K+E group: increased by 87.9%, p = 0.0352, (Figure 4B). However, the expression of tau phosphorylation at serine 396 did not significantly alter after co-administration of ethanol and ketamine for 6 months; possible inhibition was observed. After 3 months of drug treatment, only a significant increase of tau phosphorylation at serine 404 was observed in the striatum of the K30 group (increased by 128.9% compared with controls, p = 0.0074 versus controls, (Figure 4C). The fundamental increased GSK-3β level was observed in the K+E group for 6 months (increased 65%, compared with controls, p = 0.0252, (Figure 4D) with a time-dependent increase.
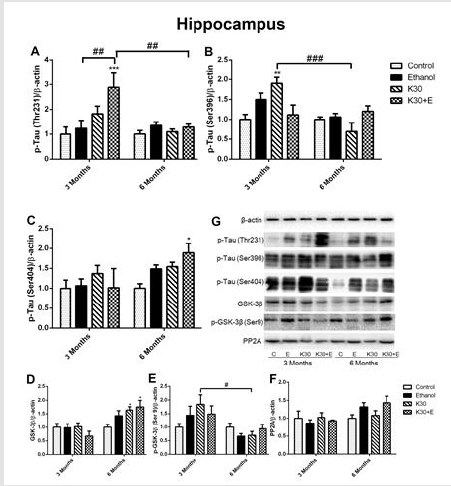
Figure 3:The effect of different ketamine treatments on the expression of tau hyperphosphorylation and related proteins in the GSK-3β signaling pathway in the hippocampus of rats (n = 4, per group). A. Quantitative analysis of p-Tau (Thr231) protein. B. Quantitative analysis of p-Tau (Ser396) protein. C. Quantitative analysis of p-Tau (Ser404) protein. D. The levels of GSK-3β was measured. E. The levels of p-GSK-3β was measured. F. The levels of PP2A was measured. G. The levels of proteins was detected by Western blot. The data was expressed as mean ± SEM. (*p < 0.05, ***p < 0.001 versus controls and #p < 0.05, ##p < 0.01, ###p < 0.001 versus drugs administration group).
The compensatory alterations of p-GSK-3β to GSK-3β were found in the striatum (Figure 4E). The expressions of p-GSK-3β were increased after 3 months of drug administration (K group: increased by 83.9%, p = 0.0372; K+E group: increased by 91.2%, p = 0.02, compared with controls, (Figure 4E) with time-dependent decrease. There was no visible alteration of PP2A protein expression observed in all drug treatment groups (Figure 4F). Different ketamine treatment enhanced Aβ, total tau, and NMDA receptors expression. We further investigated the effect of different ketamine treatments on Aβ, total tau, NMDAR1, and NMDAR2B protein expression by immunostaining. As shown in (Figure 5) and (Figure 6), for 3 months of drug administration groups, ketamine treatment induced significant increases of Aβ, total tau, and NMDAR2B protein levels in the hippocampal CA3 field (p < 0.0001 versus controls, (Figures 5A, 5B & 5D). Conversely, remarkably downregulated expressions of NMDAR1 were found for the ketamine with or without ethanol groups (p < 0.0001 and p < 0.001 versus controls, (Figure 5C). In the 6 months of drug administration groups, ketamine treatments caused considerable increases of Aβ and total tau (Figures 5A & 5B), p < 0.0001 versus controls), accompanied by the ascent of BACE1 mRNA transcription. An increase in the statistical significance of NMDAR1 expression was observed in the K30+E group (p = 0.0169 versus controls, (Figure 5C). Time-dependent decreased of Aβ, and total tau levels were found for the K+E group. In reverse, time-dependent increases in NMDAR1 levels were found for the same group.
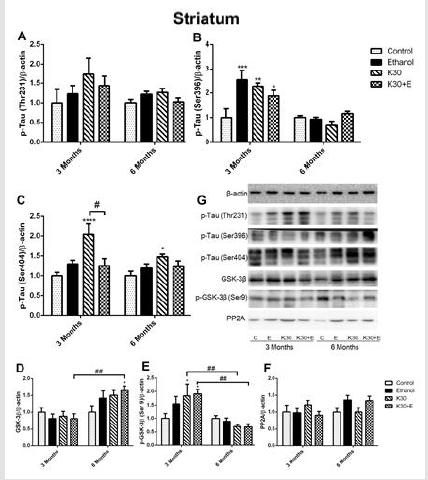
Figure 4:The effect of different ketamine treatments on the expression of tau hyperphosphorylation and related proteins in the GSK-3β signaling pathway in the striatum of rats (n = 4, per group). A. Quantitative analysis of p-Tau (Thr231) protein. B. Quantitative analysis of p-Tau (Ser396) protein. C. Quantitative analysis of p-Tau (Ser404) protein. D. The levels of GSK-3β was measured. E. The levels of p-GSK-3β was measured. F. The levels of PP2A was measured. G. The levels of proteins was detected by Western blot. The data was expressed as mean ± SEM. (*p < 0.05, **p < 0.01 versus controls and #p < 0.05, ##p < 0.01 versus drugs administration group).
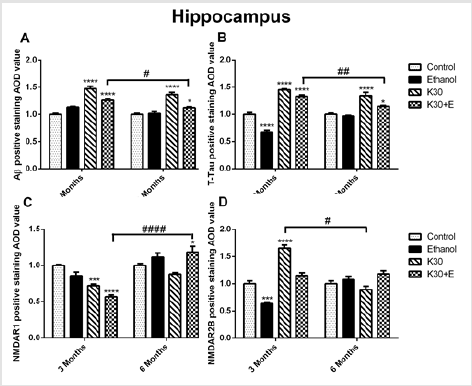
Figure 5:The effect of different ketamine treatments on Aβ, total tau, and NMDA receptors expression in the hippocampal CA3 field of rats (n = 4, per group). A. Semi-quantitative analysis of Aβ protein. B. Semi-quantitative analysis of total tau protein. C. Semi-quantitative analysis of NMDAR1. D. Semi-quantitative analysis of NMDA2B. The data was expressed as mean ± SEM. (*p < 0.05, **p < 0.01, ****p < 0.0001 versus controls and # p < 0.05, #### p < 0.0001 versus drugs administration group.
Figure 6: Protein immunostaining in CA3 from the hippocampus for four drug administration groups, shown at 3 and 6 months. Black arrows point to positive neurons, scar bar = 50 μm.
We observed the intense brown positive staining (showed with the black arrow in (Figure 7) that indicated the similar augment of Aβ and total tau in the striatum when exposure to ketamine alone and ketamine combined with ethanol for 3 months (p < 0.0001 versus controls, (Figure 8A) and (Figure 8B). In contrast to the changes in the hippocampus, ketamine with or without ethanol treatment at the same time promoted appreciable increases in and NMDAR1 protein levels (p < 0.0001, (Figure 8C). In reverse, 3 months of treatment with Ketamine and Ketamine plus ethanol were found to be effective in reducing expression of NMDAR2B (p < 0.0001 versus controls, (Figure 8D). 6 months of treatment with ketamine substantially increased expression of Aβ, total tau, and NMDAR1 (p < 0.0001 versus controls, (Figures 8A-8C). In contrast to an increase of NMDAR2B protein level in the hippocampus of the K30+E group, we observed an exceptional decrease of NMDAR2B expression in the striatum of the K30+E group (p < 0.001 versus K30 group, (Figure 8D). Additionally, we noticed that time-dependent decreased expressions of Aβ, total tau, and NMDAR1 were found for K and K+E groups, but time-dependent increased expressions of NMDAR2B were found for K and K+E groups.
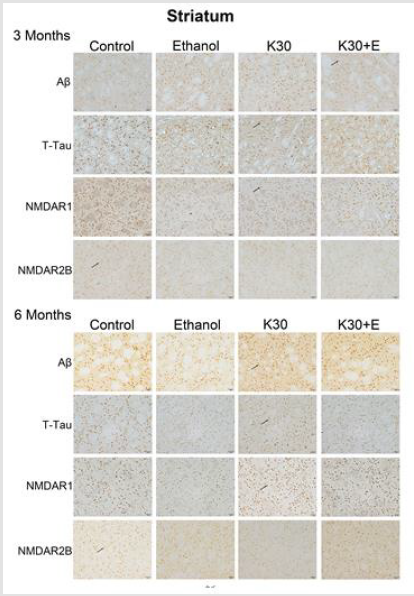
Figure 7:Protein immunostaining in the striatum for four drug administration groups, shown at 3 and 6 months. Black arrows point to positive neurons, scar bar = 50 μm.
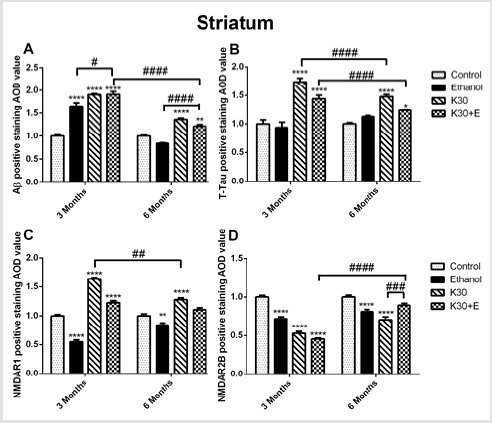
Figure 8: The effect of different ketamine treatments on Aβ, total tau, and NMDA receptors expression in the striatum of rats (n = 4, per group). A. Semi-quantitative analysis of Aβ protein. B. Semi-quantitative analysis of total tau protein. C. Semi-quantitative analysis of NMDAR1. D. Semi-quantitative analysis of NMDA2B. The data was expressed as mean ± SEM. (**p < 0.01, ***p < 0.001, ****p < 0.0001 and #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus drugs administration group.
Discussion
To better comprehend chronic ketamine users’ cognitive impairment, we considered the influence of other drugs. We explored possible synergistic interactions between ketamine and other illicit drugs on the accumulation Aβ as well as hyperphosphorylation of tau. We established rat models that treated them with ketamine only and ketamine combined with ethanol to mimic recreational ketamine abuse. Drug abusers self-administer ketamine by inhaling (60–250mg), consuming orally (200–300mg), injecting intramuscularly (75–125 mg), or injecting intravenously (50–100 mg) [23]. In this study, rat models have been treated with ketamine at the sub-anesthetic dose of 30 mg/kg. We chose 3 and 6 months as drug administration lengths according to suggested long-term drug treatment [24,25].
Effect of Different Ketamine Treatments on Tau Hyperphosphorylation and Total Tau
Hyperphosphorylated tau is the major component of the paired helical filaments (PHFs), and neurofibrillary tangles (NFTs) observed in the AD patients’ brains. 17 Tau hyperphosphorylation has a bearing on the increase in the activity of GSK-3β and Cyclin Dependent Kinase 5 (CDK5) – two enzymes involved in phosphorylating tau protein at multiple sites (Serine 396 and Serine 404) [26]. In the pre-neurofibrillary NFTs stage, the hyperphosphorylated tau locates at the serine (S) site S199, S202, and S409. In the intra-neurofibrillary NFTs stage, they locate at the S396 and threonine (T) T231. While the extra-neuronal NFTs are primarily at the S396 position [10]. In the current study, we observed significant increases of tau hyperphosphorylation at S396 and S404 in the hippocampus of rats treated with ketamine and ethanol for 6 months, accompanied by upregulation of GSK-3β mRNA transcription and protein expression. The two phosphorylated sites indicated that long-term co-administration of ketamine and ethanol probably induced hyperphosphorylated tau development to intra-neurofibrillary or extra-neuronal NFTs stage regulated through GSK-3β. The hypothesis could be further proved by the elevation of p-Tau (Ser396) found in the striatum of rats treated with long-term exposure to ketamine and ethanol mediated by GSK-3β. Serine 396 and 404 of the tau proteins locate near the microtubule – identified as essential positions involving tau-microtubules interaction in-vivo [17].
However, the alteration of GSK-3β/PP2A could not explain the significant increase of tau hyperphosphorylation at threonine (T) 231 in the hippocampus of rats treated with ketamine and ethanol for 3 months. Recently, it was reported that overexposure to methamphetamine might cause elevated phosphorylated tau proteins (S396 and T231) in the human neuroblastoma SH-SY5Y cells and in the hippocampus of SD rats, which is mediated by abnormal CDK5-regulated tau phosphorylation [27]. Additionally, tau phosphorylated at threonine 231 differentiated between AD and frontotemporal dementia. 11 Interestingly, we found a significant increase in tau phosphorylated expression at serine 404 after treating rats with Ketamine 30 mg/kg for short and long-term, which was probably mediated mutually by GSK-3β and CDK5 but needed to be further clarified. To our knowledge, this was the first report on ketamine-induced alteration of serine 404 sites of tau phosphorylation in the striatum. As the activity of GSK-3β is prevented via the phosphorylation of GSK-3β at Ser 9, [28] the reverse alterations of p-GSK-3β (Ser9), compared with GSK-3β, were observed in the hippocampus and striatum of all current rat models. PP2A balances tau phosphorylation directly via dephosphorylation of tau and indirectly via the upregulation of GSK-3β in the brain [29]. In the current study, we found a significant increase of phosphorylated tau protein at Ser 396 accompanied by upregulation of GSK-3β and PP2A expression and inhibited the activity of p-GSK-3β (Ser 9) in the hippocampus of the rats treated with ketamine and ethanol for 6 months.
The alterations probably can be explained that GSK-3β increased PP2A activity via methylation of PP2A – in turn, dephosphorylated GSK-3β. Furthermore, Ser 404 and Ser 396, among tau phosphorylation sites, were the least favorable sites for PP2A in vitro [29,30]. Tau proteins have been believed as promising candidate biomarkers for Alzheimer-type neurodegenerations. There are six different tau isoforms produced from a single gene in the adult human brain with high heterogeneity [11]. The increases in total tau and/or phosphorylated tau protein levels in cerebrospinal fluid are thought to be associated with neuronal cell death, releasing tau-related proteins into the extracellular cerebrospinal fluid [31]. We noticed that ketamine with or without ethanol treatment promoted elevated total tau protein expressions in the hippocampus and striatum, which tended to neurotoxicity.
Effect of Different Ketamine Treatments on Aβ Deposits
Aβ induces tau pathology in AD, [32] the pathological accumulation of Aβ indicates an imbalance between Aβ biosynthesis and clearance. Ketamine may reduce Aβ degradation by suppressing neprilysin expression in primary cultured astrocytes [13]. The current study supports the mechanism by which we observed extreme elevation of Aβ deposition in the hippocampal CA3 field and striatum of rats with different ketamine treatments. Significantly, co-administration of ketamine with ethanol for 3 months promoted the most considerable increase of Aβ expression in the striatum (increased by 91.4%), accompanied by the elevation of BACE1 mRNA transcription. BACE1 is the crucial enzyme that initiates Aβ accumulation, and the activity of BACE1 is the ratelimiting step in APP processing to generate Aβ. 28 However, the inhibitions of BACE1 in the hippocampus and striatum of rats with different ketamine treatments for 6 months were insufficient to explain the increased Aβ levels fully. In this study, 6 months of drug administration probably promoted the other membrane protein activity involved in Aβ production, such as presenilins 1 and 2, [33], which remains mostly elusive.
Effect of Different Ketamine Treatments on Nmda Receptors
NMDAR antagonists – both ketamine and ethanol – inhibited the degradation of exogenous Aβ via possible suppression of expression of neprilysin (NEP), the main Aβ degradation protease. The over-inhibition of NMDAR activity may also result in neuronal degeneration or death [13]. The level of NMDAR1 subunit mRNA decreased in selective regions of the hippocampus and adjacent cortical areas of AD brains. 9 Reductions in NMDAR1 expressions in the CA3 from the hippocampus of different ketamine treatment groups (3 months) confirmed this hypothesis. Contrary to the results found in AD cases, different ketamine treatment for longterm promoted appreciable increases of NMDAR1 expression in the striatum of rats. We hypothesized that repeated ketamine treatments or ketamine and ethanol co-treatment induced compensatory upregulation of NMDAR expression that allowed the accumulation of toxic levels of intracellular Ca2+, leading to neurodegeneration [8]. It also could give a better understanding of the increase in NMDAR1 expression in the CA3 from the hippocampus of the K+E group (6 months). Ketamine exposed for 3 months stimulated NMDA2B overexpression in the hippocampus, complying with the published results that promoting adult hippocampal neurogenesis by ketamine is ventral dominant via elevation of GluN2B expression [34].
Conclusion
The present study demonstrates that region-specific AD-related biological events may damage the brain induced by recreational ketamine abuse. There are still limitations of the proposed possible mechanisms of tau hyperphosphorylation and extracellular Aβ deposits that need to be explored in detail. Our study might open a new scenario for the molecular mechanism of cognitive dysfunction induced by recreational ketamine abuse by drawing on the AD investigation’s achievements.
Declarations
Funding
This work has been supported by the National Natural Science Foundation of China (No. 81460213), Yunnan Applied Basic Research Projects-Joint Special Project (No. 2017FE468 (-020)), and Kunming Medical University Creative Group Programme (No. CXTD201604).
Conflicts of Interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Consent for Publication
Not applicable
Availability of Data and Material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request
Authors’ Contributions
All authors participated in the design, interpretation of the studies, and analysis of the data and review of the manuscript; LZ and PQ conducted the experiments, HL, SH and JG performed the statistical analysis, created figures and tables. JM revised the manuscript. HL and RY designed the study and drafted the manuscript. The authors declare that all data were generated inhouse and that no paper mill was used.
References
- Krystal JH, Karper LP, Seibyl JP, GK Freeman, R Delaney, et al. (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51(3): 199-214.
- Liao Y, Tang YL, Hao W (2017) Ketamine and international regulations. Am J Drug Alcohol Abuse 43(5): 495-504.
- Zhang C, Tang WK, Liang HJ, Gabor Sandor Ungvari, Shih Ku Lin, et al. (2018) Other drug use does not impact cognitive impairments in chronic ketamine users. Drug Alcohol Depend 186: 1-8.
- Curran HV, Morgan C (2000) Cognitive, dissociative and psychotogenic effects of ketamine in recreational users on the night of drug use and 3 days later. Addiction 95(4): 575-590.
- Turktan M, Yilmaz MB, Hatipoglu Z, Seda Ilgaz, Esma Deniz Barc, et al. Molecular determinants of behavioral changes induced by neonatal ketamine and dexmedetomidine application. Journal of neural transmission 126(12): 1577-1588.
- Gerlai R, McNamara A (2000) Anesthesia induced retrograde amnesia is ameliorated by ephrinA5-IgG in mice: EphA receptor tyrosine kinases are involved in mammalian memory. Behav Brain Res 108: 133-143.
- Mickley GA, Remmers Roeber DR, Crouse C (2000) Ketamine blocks a taste recognition memory in fetal rats. Pharmacol Biochem Behav 67(3): 575-581.
- Xu K, Lipsky RH (2015) Repeated ketamine administration alters N-methyl-D-aspartic acid receptor subunit gene expression: Implication of genetic vulnerability for ketamine abuse and ketamine psychosis in humans. Exp Biol Med (Maywood) 240(2): 145-155.
- Ulas J, Cotman CW (1997) Decreased expression of N-methyl-D-aspartate receptor 1 messenger RNA in select regions of Alzheimer brain. Neuroscience 79(4): 973-982.
- Yeung LY, Wai MS, Fan M, YT Mak, WP Lam, et al. (2010) Hyperphosphorylated tau in the brains of mice and monkeys with long-term administration of ketamine. Toxicol Lett 193(2): 189-193.
- Hampel H, Blennow K, Shaw LM, Yvonne C Hoessler, Henrik Zetterberg, et al. (2010) Total and phosphorylated tau protein as biological markers of Alzheimer's disease. Exp Gerontol 45(1): 30-40.
- Chang Y, Li JY, Jayakumar T, Shou Huang Hung, Wei Cheng Lee, et al. (2017) Ketamine, a Clinically Used Anesthetic, Inhibits Vascular Smooth Muscle Cell Proliferation via PP2A-Activated PI3K/Akt/ERK Inhibition. International journal of molecular sciences 18(12): 2545.
- Yamamoto N, Arima H, Naruse K, Hideo Taniura, Hiroyuki Hirate, et al. (2013) ketamine reduces amyloid beta-protein degradation by suppressing neprilysin expression in primary cultured astrocytes. Neuroscience letters 545: 54-58.
- Glabe CC (2005) Amyloid accumulation and pathogensis of Alzheimer's disease: Significance of monomeric, oligomeric and fibrillar Abeta. Sub-cellular biochemistry 38: 167-177.
- Liu Q, Xu TY, Zhang ZB, Chi Kwan Leung, Ding Yun You, et al. (2017) Effects of co-administration of ketamine and ethanol on the dopamine system via the cortex-striatum circuitry. Life Sci 179: 1-8.
- Ren SQ, Yao W, Yan JZ (2018) Amyloid beta causes excitation/inhibition imbalance through dopamine receptor 1-dependent disruption of fast-spiking GABAergic input in anterior cingulate cortex. Sci Rep 8: 302.
- Elyaman W, Terro F, Wong NS (2002) In vivo activation and nuclear translocation of phosphorylated glycogen synthase kinase-3beta in neuronal apoptosis: Links to tau phosphorylation. Eur J Neurosci 15: 651-660.
- Peters F, Salihoglu H, Rodrigues E, Etienne Herzog, Tanja Blume, et al. (2018) BACE1 inhibition more effectively suppresses initiation than progression of beta-amyloid pathology. Acta Neuropathol 135(5): 695-710.
- Wai MS, Chan WM, Zhang AQ (2012) Long-term ketamine and ketamine plus alcohol treatments produced damages in liver and kidney. Hum Exp Toxicol 31(9): 877-886.
- Tan S, Chan WM, Wai MS, Lawrence KK Hui, Vivian WK Hui, et al. (2011) Ketamine effects on the urogenital system--changes in the urinary bladder and sperm motility. Microsc Res Tech 74(12): 1192-1198.
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4): 402-408.
- Jin H, Hu Z, Dong M, Yidong Wu, Zhirui Zhu, et al. (2013) Ketamine induces tau hyperphosphorylation at serine 404 in the hippocampus of neonatal rats. Neural Regen Res 8(17): 1590-1596.
- Kalsi SS, Wood DM, Dargan PI (2011) The epidemiology and patterns of acute and chronic toxicity associated with recreational ketamine use. Emerg Health Threats J 4: 7107.
- Shen CH, Wang SC, Wang ST, Shu Mei Lin, Jiann Der Wu, et al. (2016) Evaluation of urinary bladder fibrogenesis in a mouse model of long-term ketamine injection. Mol Med Rep 14(3): 1880-1890.
- Sun L, Lam WP, Wong YW, LH Lam, HC Tang, et al. (2011) Permanent deficits in brain functions caused by long-term ketamine treatment in mice. Hum Exp Toxicol 30(9): 1287-1296.
- Busceti CL, Biagioni F, Riozzi B (2008) Enhanced tau phosphorylation in the hippocampus of mice treated with 3,4-methylenedioxymethamphetamine ("Ecstasy"). J Neurosci 28(12): 3234-3245.
- Xiao N, Zhang F, Zhu B, Chao Liu, Zhoumeng Lin, et al. (2018) CDK5-mediated tau accumulation triggers methamphetamine-induced neuronal apoptosis via endoplasmic reticulum-associated degradation pathway. Toxicol Lett 292: 97-107.
- Ali T, Yoon GH, Shah SA, Hae Young Lee, Myeong Ok Kim, et al. (2015) Osmotin attenuates amyloid beta-induced memory impairment, tau phosphorylation and neurodegeneration in the mouse hippocampus. Sci Rep 5: 11708.
- Qian W, Shi J, Yin X, Khalid Iqbal, Inge Grundke Iqbal, et al. (2010) PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J Alzheimers Dis 19(4): 1221-1229.
- Wang Y, Yang R, Gu J, Xiaomin Yin, Nana Jin, et al. (2015) Cross talk between PI3K-AKT-GSK-3beta and PP2A pathways determines tau hyperphosphorylation. Neurobiology of aging 36(1): 188-200.
- Gilman S, Koller M, Black RS, SG Griffith, NC Fox, et al. (2005) Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64(9): 1553-1562.
- Ittner LM, Ke YD, Delerue F, Mian Bi, Amadeus Gladbach, et al. (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142(3): 387-397.
- Villamil Ortiz JG, Barrera Ocampo A, Piedrahita D (2016) BACE1 RNAi Restores the Composition of Phosphatidylethanolamine-Derivates Related to Memory Improvement in Aged 3xTg-AD Mice. Front Cell Neurosci 10: 260.
- Yamada J, Jinno S (2019) Potential link between antidepressant-like effects of ketamine and promotion of adult neurogenesis in the ventral hippocampus of mice. Neuropharmacology 158: 107710.
