 Case Report
Case ReportBackground
Almost a total of 1317 Hb variants have been identified (HbVar
database) [1], the four most common worldwide are Hb S, Hb E, Hb
C, and Hb D, in the order of decreasing prevalence [2]. Haemoglobin
(Hb) E is the most prevalent variant in Southeast Asia (Thailand,
Myanmar, Cambodia, Laos, Vietnam), where its prevalence is 30-
60% [3-6]. The prevalence of HbE in India is about 3.5% with an
increased clustering in Kolkata (22%) and Assam (50-80%) [7]. Hb
E results from a G→A substitution in codon 26 of the β globin gene.
This produces an abnormal Hb (glutamate is replaced by lysine)
and activates a cryptic splice site at codon 25-27 of the β-globin
gene, resulting consequently in abnormal processing for messenger
RNA (mRNA). The level of normally spliced mRNA become reduced
and because a new stop codon is generated, the abnormally spliced
mRNA become nonfunctional [7,8].
Fortunately, only a minor activation of the alternative splicing
pathway is associated with this mutation resulting in a moderate
reduction of the normally spliced βE globin mRNA 72. Hb E trait
(E heterozygosity; ββE) and Hb E disease (E homozygosity; βEβE)
are mild disorders. Although the Hb E mutation alone does not
cause any significant clinical problems, its interactions with
various forms of α and β thalassemia produce a very wide range of
clinical syndromes of varying severity [8,9]. Different phenotypes
could be noticed with the compound heterozygote state of Hb
Eβ-thalassemia ranging from a complete lack of symptoms to
transfusion dependency [3,7,8,10]. Experiments were carried out
in vitro at temperatures ranging from 38 to 41°C showed that there
was a mild instability of Hb E but there is no evidence that this is
the case in vivo [9-14]. It is noticeable that the E allele causes a mild
thalassemia, while Eβ0 thalassemia shows severe phenotypes. This
marked paradox in phenotypes could not be fully explained up till
now. It is reported that HbE is sensitive to oxidative stress. Does
this or other properties of HbE explain the variable severity of the
Eβ0 thalassemia? This question is still waiting for an answer [11].
The aim of this report is to present a case of Hb E homozygosity
discovered accidentally trying to cast shadow on a mutation not
prevalent in the Middle East.
Case Report
A Consent has been taken from a 26-year-old male, from
Kolkata, India, came to Najran University Hospital, Saudi Arabia for
routine investigation. He did not complain from anemia or receive
treatment. He gave a history of hemolytic attack because of high
fever indifintily due to infection with malaria and only one blood
transfusion. On examination, there were no significant clinical
findings such as organomegaly, icterus, or thalassemic bone changes.
CBC were carried out using Sysmex XS 500i (Sysmex, https://www.
sysmex.com/). It showed mild microcytic hypochromic anaemia with increase in RBC distribution width (RDW-CV) and eosinophilia
(Table 1). Peripheral blood smear showed frequent target cells, and
spherocytes as shown in (Figure 1). Serum biochemical analysis
were carried out using COBAS C311 (Roche, https://www.roche.
com/). Results were normal for liver and kidney functions except
for mild increase in bilirubin (1.39 mg/dL, mostly indirect of 0.98
mg/dL).
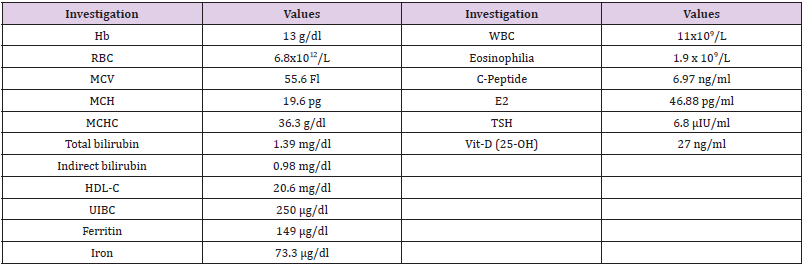
Table 1: Some hematological parameters of the reported case.
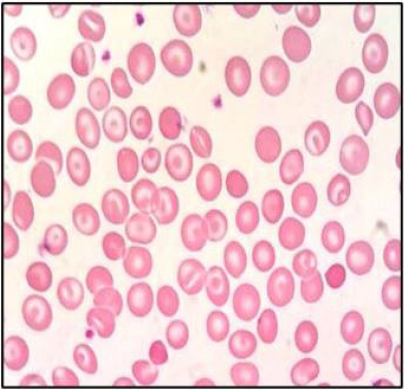
Figure 1: Peripheral blood smear of the reported case.
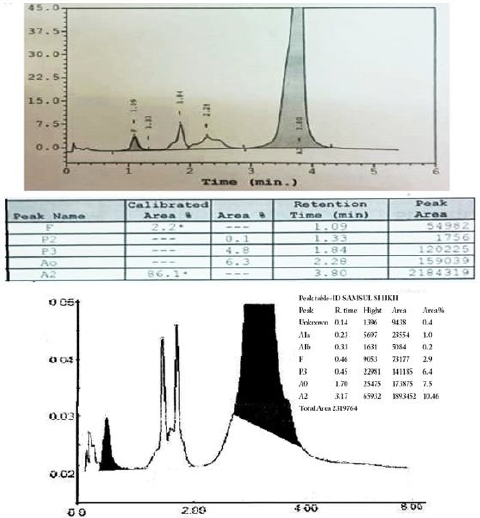
Figure 2: HPLC of the reported case: a. D-10 instrument. b. Variant II instrument.
Discussion
Hb E has two compartments; one is the Hb E (α2βE2) itself
which behaves like a rather normal Hb in normal conditions. The
other one is the thalassemic compartment with excess α globin
chain leading to a mild haemolytic anaemia .Considering history,
ethnic origin, clinical findings and laboratory findings of the
patient, the diagnosis was consistent with Hb E homozygosity. He
showed a very mild, clinically asymptomatic, hemolytic microcytic
hypochromic anemia with many target cells on peripheral blood
smear [9]. The presence of an abnormal peak of around 85% in
the A2 window with different retention time than Hb A2 (usually
around 3.2 min), a mild increase in Hb F (Figure 2), a mild increase
in the unconjugated bilirubin (Table 1), indicates the mildness
of the pathophysiology of the thalassemic compartment of the
disease. The presence of small peaks on HPLC was explained by
post-translational modification of Hb E13 . Hb E masked the Hb
A2 on HPLC, hense we could not configure its percentage which is
especially important in the diagnosis of δβ thalassemia. We noticed
the presence of many spherocytes in the peripheral blood smear
with an increase of MCHC. Similar findings occur in Hb C due an
increase in the activity of K:Cl- cotransport that induces the loss of
K+ and subsequently of intracellular water [15].
The patient had a low HDL-C, a slightly increased E2 and
TSH hormones, a slightly increased C-peptide which could not be
explained. The patient gave a history of high fever, indifintly due to
malaria infection, with a hemolytic attack and a blood transfusion
once, which could match the published of the instability of Hb E in
high fever [14]. Although investigation for malaria was negative, yet
the presence of eosinophilia (Table 1) may be due to an old infection
[16]. The main differential diagnosis is the Eβ0 thalassemia. Hb E is
usually around 50% with severe clinical presentation and marked
increase in Hb F [17]. Eβ+ thalassemia is milder and characterized
by the presence of Hb A. As there are only two β globin genes, one
on each chromosome, usually the majority of the Hb is abnormal in
case of homozygous β mutation. Other β globin chain variants elute
in the A2 window on HPLC are D-Iran, Osu Christiansborg, Deer
Lodge, G-Coushatta, G-Copenhagen, D-Ouled Rabah, Ocho Rios,
Korle-Bu, G-Ferrara, Zürich and Rocky Mountain, Hb Abruzzo and
M-Saskatoon). Mostly they are non-pathogenic with normal or near
normal clinical presentation. Two fusion gene mutations (Lepore
and Kenya) also elute in the A2 window.
Hb Lepore, a δβ fusion gene is another thalassemic variant result
in δβ thalassemia. In homozygosity, it gives severe thalassaemia
intermedia to thalassaemia major with HbF 80%, Hb Lepore 20%
and no Hb A or Hb A2 [18]. Hb Kenya (considered an HPFH), a
gamma beta (Aγβ) hybrid with expected 22.5 kb deletion is also
expressed at low percentage (6-23 %) with increased Hb F (5-28%)
in heterozygotes. Four alpha globin chain mutations also elute in the
A2 window (Spanish Town, G-Honolulu, Toulon and Fort Worth).
They are expressed at low percentage (11-21% in heterozygous)
usually with no clinical manifestation. As an α-variant, they show the
presence of the characteristic minor Hb A2 variant peak (α2M δ2)
immediately after the α variant peak, which is missing in β variants.
Many other Hb variants (Alabama, Abington, Akron, Bethesda,
Chandigarth, Denver, Ethiopia, G Galveston, G Taipei, Hoshida,
Hamadan, Jeddah, Loves Park, Muravera, Nebraska, San Bruno,
Santa Juana, SId (the aged adduct of Hb S due to glutathione) and
Tubingen) can elute between the A and the A2 windows on HPLC
masking the Hb A2 [19].The majority of hemoglobinopathy present
in the western and eastern provinces of Saudi Arabia, particularly
in the southwestern province. Najran city, in the south of KSA, had
the least prevalence of haemoglobinopathies (β Thalassemia trait
(2.4 %), zero β thalassemia disease, 12.5% sickle carrier and 0.3%
sickle disease). This in contrast to Jazan, also a south city, in which
haemoglobinopathies are clustering. Few cases of Hb E may be
present in Najran and many must be present in Jazan, but there is
no report about the prevelance as far as we know. Orientation by
the different phenotypes by Saudi doctors is important to prevent
misdiagnosis in haemoglobinopathy. Orientation by different
genotypes is critical in premarriage consultation and prenatal
screening programs in the kingdom.
References
- (2014) HbVar database: A Database of Human Hemoglobin Variants and Thalassemia mutations.
- Yedla N, Kuchay MS, Mithal A (2015) Hemoglobin E disease and glycosylated hemoglobin. Indian J Endocr Metab 19(5): 683-685.
- Olivieri NF, Pakbaz Z, Vichinsky E (2011) Hb E/beta-thalassaemia: a common & clinically diverse disorder. Indian J Med Res 134(4): 522-531.
- Kohne E (2011) Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Dtsch Arztebl Int 108(31-32): 532-540.
- Katsanis E, Luke KH, Hsu E, Yates JR (1987) Hemoglobin E: a common hemoglobinopathy among children of Southeast Asian origin. CMAJ 137(1): 39-42.
- Little RR, Roberts WL (2009) A review of variant hemoglobins interfering with hemoglobin A1c measurement. J Diabetes Sci Technol 3(3): 446-4451.
- Weatherall DJ (2008) Hemoglobinopathies worldwide: present and future. Curr Mol Med 8(7): 592-599.
- Gibbons R, Higgs DR, Olivieri NF, Wood WG (2001) The β and δβ thalassaemias in association with structural haemoglobin variants. In: Weatherall DJ, Clegg JB, (Eds.), The Thalassaemia Syndromes (4th edn). Oxford, United Kingdom: Blackwell Science pp. 393-449
- Fucharoen S1, Weatherall DJ (2012) The hemoglobin E thalassemias. Cold Spring Harb Perspect Med 2(8): a011734.
- Fucharoen S, Ketvichit P, Pootrakul P, Siritanaratkul N, Piankijagum A, et al. (2000) Clinical manifestation of betathalassemia/hemoglobin E disease. J Pediatr Hematol Oncol 22(6): 552-557.
- Acquaye JK, Omer A, Ganeshaguru K, Sejeny SA, Hoffbrand AV (1985) Non-benign sickle cell anaemia in western Saudi Arabia. Br J Haematol 60(1): 99-108.
- Memish ZA, Saeedi MY (2011) Six-year outcome of the national premarital screening and genetic counseling program for sickle cell disease and beta-thalassemia in Saudi Arabia. Ann Saudi Med 31(3): 229-235.
- Weatherall DJ, Clegg JB (2001) Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ 79(8): 704-712.
- Rees DC, Clegg JB, Weatherall DJ (1998) Is hemoglobin instability important in the interaction between hemoglobin E and b thalassemia? Blood 92(6): 2141-2146.
- Nagel RL, Fabry ME, Steinberg MH (2003) The paradox of hemoglobin SC disease. Blood Rev 17(3): 167-178.
- Saidu AY, Sadiya H, Mustapha UK, Kumurya AS, Fana SA, et al. (2015) Pathophysiology of Eosinophilia in Malarial Infection in Patients Attending Usmanu Danfodiyo University Teaching Hospital (Uduth) Sokoto, Nigeria. IOSR Journal of Dental and Medical Sciences 14(10): 105-110.
- Vichinsky E (2007) Hemoglobin E syndromes. Hematology Am Soc Hematol Educ Program pp. 79-83.
- Stephens AD, Angastiniotis M, Baysal E, Chan V, Fucharoen S, et al. (2012) ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol 34(1): 1-13.
- Alsaeed ES, Farhat GN, Assiri AM, Memish Z, Ahmed EM, et al. (2018) Distribution of hemoglobinopathy disorders in Saudi Arabia based on data from the premarital screening and genetic counseling program, 2011-2015. J Epidemiol Glob Health 7(Suppl 1): S41-S47.
