 Mini Review
Mini ReviewAbstract
Protein folding is a big challenge in life science post-gene era. Particularly, a larger number of sequences have been well known but most of them still lack 3D structural information. A novel approach has been developed to overcome the hurdle. Based on the folded of 5 amino acids, the protein folding conformation for any protein 3D structure can be complete described by Protein Folding Shape Code (PFSC). Furthermore, all possible folding shapes for 5 amino acids were gathered and expressed in PFSC as digitized description. Finally, along a protein sequence from N-terminate to C-terminate, the folding variations can be presented in Protein Folding Variation Matrix (PFVM), and then possible conformations can be assembled.
Keywords: PDB: Protein Data Bank; UniProt: Universal Protein Resource; PFSC: Protein Folding Shape Code; PFVM: Protein Folding Variation Matrix
Introduction
Rich protein structural data has been cumulated in sequences with one-dimensional information and in conformations with 3D structures. A large number of protein sequences are available in database today. To determine the order of amino acids, some of protein sequences may be measured by LC-MS experiments. However, a huge number of protein sequences can be directly acquired by transaction of DNA and then translation of mRNA following genome development. Over 167,000,000 protein sequences have been accumulated in Universal Protein Resource (UniProt), and about 560,000 sequences among them were manually annotated. Furthermore, the knowledge of conformations is significant for biology functions because the protein folding plays important role as well as sequence for protein functions. The protein folding conformation can be straightly determinate according given 3D structural data. The protein 3D structures may be determined by either experimental measurements or computational approaches. Experimentally, the protein structures can be measured by Nuclear Magnetic Resonance (NMR), X-ray crystallography or Transmission Electron Cryomicroscopy (CryoTEM), and so far over 155,000 of 3D structure data have been available in Protein Data Bank (PDB). However, only less than 0.5% of proteins have 3D structural data relative to hundreds of millions of protein sequences. Also, it is impossible to keep up with the pace of increase of number of protein one-dimensional sequences. On the other hand, the computational approaches become an important methodology to predict the protein 3D structures. So far, various methodologies for protein structure predictions have been developed [3-16]. Since 1994, the platform of Critical Assessment of techniques for protein Structure Prediction (CASP) has provided a worldwide platform to promote the development of protein structure predictions. Overall, the protein structure prediction still cannot to resolve all proteins in the near future because of the request of vast computational resources. In recent years, the artificial intelligence with deep learning approach has been applied into protein folding. In 2018, Google’s Alpha Fold made a big progress in protein structure prediction. , Although this development, it’s still a long way for thoroughly solving the protein folding problem [1-5].
Challenges in Protein Folding Conformation
In generally, the thorough resolution for protein folding has several challenges. First, over 167,000,000 of proteins in UniProt are known only in one-dimensional sequences without 3D structural information. It is apparently to deal with such gigantic number of protein sequences is hard to be accomplished by either traditional experimental measurements or computation approaches. In contradiction, to prefer more accuracy with experiments and computational approaches would make more difficult to achieve the goal. Second, any single protein sequence may fold into an astronomical number of conformations which aggravate the task with further difficulty. In principle, the number of protein folding essentially is a function of the order of amino acids. Until now, scientists have put much effort trying to overcome the difficulty, however, the regulation or correlation between the protein folding and the order of amino acids has never been known. Third, even as an astronomical number of conformations were obtained, how to present or analyze such gigantic number of structural data is unimaginable. To face so many obstacles, it is not surprised that the protein folding was defined as one of 100 hard scientific questions in this century by Science in 2005 [6-10].
Protein Structure Fingerprint Technology
In order to handle a gigantic number of protein structural data, the protein structure fingerprint technology has been developed to overcome these hurdles. In this novel approach, all possible folds of 5 amino acids are covered by Protein Folding Shapes Code (PFSC) as 27 alphabetical letters. Then, any protein conformation can be completely described by a PFSC string. A database of 5AAPFSC is created to gather all possible folds in PFSC for 5 amino acids. Consequently, the complete local folding variations can be obtained according the sequence and assemble in Protein Folding Variation Matrix (PFVM), which contains local folding variations along sequence. In addition, the protein conformations in an astronomical number as well as the most possible conformations can be constructed with PFVM [11-16].
The procedure to obtain PFVM is briefly illustrated in Figure 1, i.e. with one-dimensional sequence as the input, the comprehensive protein folding variations are interpreted by PFSC, and then the PFVM can be obtained as the output. With digitized description, the PFVM holds the gigantic folding variations at a glance. Here, human protein of small cell adhesion glycoprotein (SMAGP_HUMAN) is taken as an example to demonstrate the feature of the protein structure fingerprint technology. Actually, the 3D structure of protein SMAGP_HUMAN is unknown. However, its sequence with 97 amino acids is available from UniProt database. To access on-line service (www.micropht.com) and perform the function, the PFVM of protein SMAGP_HUMAN was obtained, and showed in Figure 2. With combination of all folding variation in PFSC letters in PFVM, 3.55x1054 of folding conformations can be assembled. These conformations can be explicitly expressed by PFSC strings. Table 1 displayed 9 of possible conformations as well as folding images as sample results. Conclusion, the protein structure fingerprint technology is able well to reveal the relationship between the protein folding and the order of amino acids in PFVM. Also, an astronomical number of conformations of protein folding conformations can be explicitly acquired with digitized PFSC description [17-24].
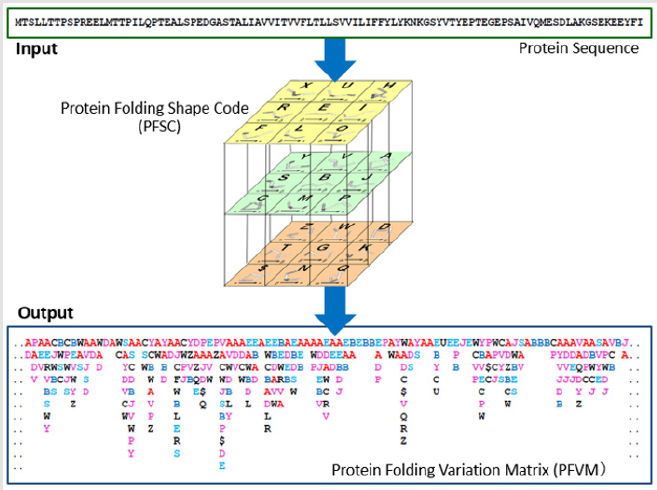
Figure 1: The procedure from sequence to protein folding variations. 27 Protein Folding Shapes Code (PFSC) is presented in the cubic. With one-dimensional sequence as the input, the Protein Folding Variation Matrix (PFVM) is obtained. The PFVM hold complete folding variations for 5 amino acids in protein.
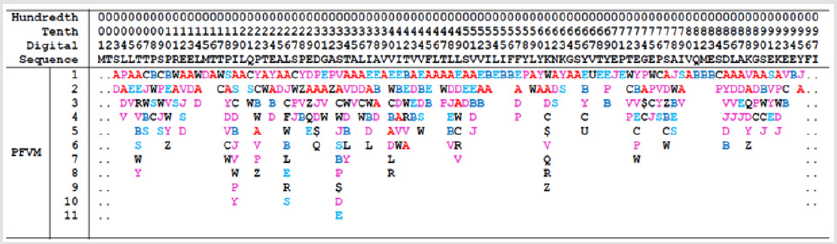
Figure 2: The Protein Folding Variation Matrix (PFVM) for human protein of small cell adhesion glycoprotein (SMAGP_ HUMAN). Top section: the numerical ruler and the sequence. Bottom section: the PFVM of SMAGP_HUMAN. The PFSC letters in each column are the local folding variations for 5 amino acids. The PFSC letters are marked by colors: red is for typical helix fold; blue for typical beta fold; pink and light blue for folds with partial helix or beta; black for irregular folds.

Table 1: Samples of folding conformations of human protein of small cell adhesion glycoprotein (SMAGP_HUMAN). Top section: the numerical ruler and the sequence. According the PFVM of SMAGP_HUMAN in Table 1, total number of possible folding conformations is 3.55 x 1054 are able to be assembled with the local folding variations. Conformations section: list 9 of possible conformations in PFSC as samples. Bottom section: display 9 of 3D images of folding conformations corresponding to the PFSC list.
Conclusion
With protein structure fingerprint technology, the protein folding variations can be directly obtained from sequence, which are well displayed by PFVM. With amino acid sequence as input, the PFVM as output will be obtained with free access on web server www.micropht.com.
References
- http://www.uniprot.org/
- https://www.rcsb.org/
- Compiani M, Capriotti E (2013) Computational and theoretical methods for protein folding. Biochemistry 52(48): 8601-8624.
- Guo JT, Ellrott K, Xu Y (2008) A historical perspective of template-based protein structure prediction. Methods Mol Biol 413: 3-42.
- Dorn M, E Silva MB, Buriol LS, Lamb LC (2014) Three-dimensional protein structure prediction: Methods and computational strategies. Comput Biol Chem 53PB: 251-276.
- Brylinski M (2015) Is the growth rate of Protein Data Bank sufficient to solve the protein structure prediction problem using template-based modeling: Bio-Algorithms and Med-Systems 11(1): 1-7.
- J Yang, R Yan, A Roy, D Xu, J Poisson, et al. (2015) The I-TASSER suite: Protein structure and function prediction. Nature Methods 12: 7-8.
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D (2007) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics Unit-5.6.
- Liu T, Tang GW, Capriotti E (2011) Comparative modeling: The state of the art and protein drug target structure prediction. Comb Chem High Throughput Screening 14: 532-537.
- Yang L, Tan CH, Hsieh MJ, Wang J, Duan Y (2006) New-generation amber united-atom force field. J Phys Chem B 110: 13166-13176.
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, et al. (2009) CHARMM: The biomolecular simulation program. J Comput Chem 30: 1545-1614.
- Riniker S, Christ CD, Hansen HS, Hünenberger PH, Oostenbrink C (2011) Calculation of relative free energies for ligand-protein binding, solvation, and conformational transitions using the GROMOS software. J Phys Chem B 115: 13570-13577.
- Honig B (1999) Protein folding: From the levinthal paradox to structure prediction. J Mol Biol 293: 283-293.
- Onuchic JN, Wolynes PG (2004) Theory of protein folding. Curr Opin Struct Biol 14: 70-75.
- Vallat B, Madrid-Aliste C, Fiser A (2015) Modularity of protein folds as a tool for template free modeling of structures. PLoS Comput Biol 11(8): e1004419.
- Zhang J, Li W, Wang J, Qin M, Wu L, et al. (2009) Protein folding simulations: From coarse-grained model to all-atom model. IUBMB Life 61: 627-643.
- Moult J, Fidelis K, Kryshtafovych A, Tramontano A (2011) Critical assessment of methods of protein structure prediction (CASP): Round IX. Proteins 79(Suppl. 10): 1-5.
- Benedix A, Becker CM, de Groot BL, Caflisch A, Bockmann RA (2009) Predicting free energy changes using structural ensembles. Nat Methods 6: 3-4.
- https://deepmind.com/blog/alphafold/
- Evans R, Jumper J, Kirkpatrick J, Sifre L, Green TF G, et al. (2018) De novo structure prediction with deep-learning based scoring In: Thirteenth critical assessment of techniques for protein structure prediction (Abstracts) p. 1-4.
- Levinthal C (1969) How to fold graciously. In Mossbauer ̈ spectroscopy in biological systems. Allerton House, Monticello, IL, p. 22-24.
- Cobb M (2017) 60 years ago, Francis crick changed the logic of biology. Plos Biology 15(9): e2003243.
- Dill KA, Maccallum JL (2012) The protein-folding problem, 50 years. Science 338(6110): 1042-1046.
- Yang J (2008) Comprehensive description of protein structures using protein folding shape code. Proteins 71(3): 1497-1518.
